Case Report:
Patient:
Mr. X, 99 years old
History:
A 99-year-old patient was admitted to the hospital for complaints of decrease in response and blood in urine.
Past history:
Patient is a known case of benign prostatic hyperplasia/ subdural hepatoma-s/p evacuation.
Clinical Presentation:
On admission, Mr. X was drowsy .Vital signs were unremarkable. Neurological examination revealed E4V2M3. Other systemic examinations were normal.
Investigations:
-
- -Blood Tests : Low hemoglobin, elevated WBC(3,31,000), C-reactive protein (CRP), urea and creatinine
- – Peripheral smear :
-
- RBC- Macrocytic hypochromic RBC. Anisocytosis with tear drop cells and microspherocytes.
- WBC- Markedly increased in number, with DC: Neutrophils 37% Lymphocytes 02% Monocytes 01% Eosinophils 01%
-
- Basophils 01% Blast 07% PMC 09% MC 25% MMC 04% Band 13% NRBC 3/100 WBC.
- – BCR- ABL1transcript : Not detected
- – OncohaemNGS PANEL : PATHOGENIC MUTATION DETECTED in ASXL1, EZH2, STAG2, NRAS and KRAS genes
- – UrineAnalysis: Plenty of pus cells, protein 4+, blood 2+.
- – Urineculture grew Proteus
- – Imaging:
- CT KUB: Bladder diverticula, prostatomegaly, moderate splenomegaly, focal hyperdensity in the bladder base
- CT Chest: Bilateral pleural effusions, cardiomegaly, honeycombing in the right lower lobe’s basal aspect, fibrotic band in the left upper
- – Echocardiogram: Sclerotic aortic valve with mild aortic regurgitation, left ventricular hypertrophy, mild mitral regurgitation, mild tricuspid regurgitation, mild pulmonary artery hypertension, normal cardiac chamber sizes, and systolic No pericardial effusion or LV thrombus.
-
Treatment:
He was started on hydroxyurea after consultation with Hematologist. Repeat blood investigations showed wbc in decreasing trend.
| WBC | |
| Day 0 | 3,31,000 |
| Day 2 | 3,14,000 |
| Day 4 | 2,20,000 |
| Day 9 | 13,750 |
However, the patient’s condition deteriorated, leading to the initiation of high-flow nasal cannula and noradrenaline due to septic shock, and he was subsequently declared deceased.
Discussion:
Chronic myeloid leukemia (CML) represents roughly 14% of all leukemias and primarily affects adults between 40 and 60 years old. It is driven by the Philadelphia chromosome, resulting from a balanced reciprocal translocation between chromosomes 9 and 22. This genetic alteration generates the BCR-ABL1 chimeric oncogene , which encodes a tyrosine kinase protein with a molecular weight of 210 kDa that leads to unchecked cell proliferation and impaired apoptosis. The BCR-ABL1 oncoproteins is expressed in various hematopoietic cells (myeloid, erythroid, megakaryocytes, and monocytes; less often mature B lymphocytes; rarely mature T lymphocytes )but not in other body cells.
Common symptoms, when they occur, typically involve signs of anemia and splenomegaly. These may include fatigue, malaise, weight loss (with a high leukemia burden), or early satiety, left upper quadrant pain, or masses due to splenomegaly. Less common symptoms can result from thrombotic or hyperviscosity-related complications due to severe leukocytosis or thrombocytosis, such as priapism, cardiovascular issues, myocardial infarction, venous thrombosis, visual disturbances, dyspnea, pulmonary insufficiency, drowsiness, loss of coordination, confusion, or cerebrovascular accidents.
CML typically progresses through an indolent chronic phase, followed by an accelerated phase, and eventually a terminal blastic phase. The blast crisis is marked by increased blast cells in the bone marrow or blood and leukemic infiltrates in soft tissues or skin. Unlike acute leukemias, which can quickly reverse or become fatal, CML has a slower progression. Without initial cure, it may lead to acute leukemia (75% myeloid, 25% lymphoid) or myelofibrosis, with a median survival of 3–4 years.
Diagnostic investigations involve blood counts, bone marrow aspirates, and cytogenetic analysis. Typical findings include leukocytosis commonly ranging from 10–500 × 10^9/L. The peripheral blood differential typically shows a left shift in hematopoiesis with a predominance of neutrophils and the presence of bands, myelocytes, metamyelocytes, promyelocytes, and blasts (usually ≤5%). Increased basophils and/or eosinophils are also frequent. While thrombocytosis is common, thrombocytopenia is rare and, if present, may indicate a worse prognosis or disease acceleration. Biochemical abnormalities include low leukocyte alkaline phosphatase levels and elevated vitamin B12, uric acid, lactic dehydrogenase, and lysozyme. The bone marrow typically shows hypercellularity with significant myeloid hyperplasia and a myeloid-to-erythroid ratio of 15–20:1. Marrow blasts are usually 5% or less; higher levels indicate a worse prognosis or possible transformation to an accelerated phase if ≥15%.
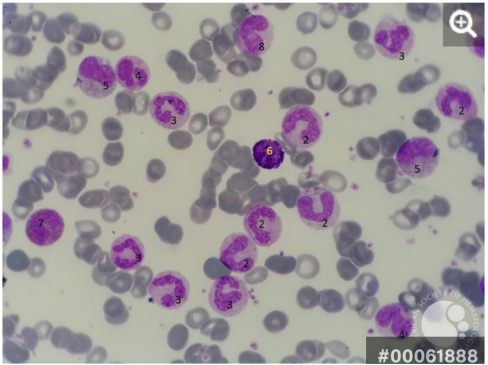
Peripheral smear of a CML patient
1- Blast cell (probably basophilic normoblast)
2- Band cell
3- Band cells going to be segmented
4- Dysplastic cell
5- Metamyelocyte
6- Basophil
7- Eosinophilic myelocyte
8- Latemeta myelocyte
Diagnostic confirmation of CML typically involves detecting the BCR-ABL1 fusion gene. In some cases, this may not be apparent with standard karyotyping but can be identified using fluorescence in situ hybridization (FISH) or polymerase chain reaction (PCR). Somatic mutations may help to diagnose but are not specifically pathognomonic of the disease, with the most detected including ASXL1, SETBP1, NRAS, KRAS, SRSF2, and TET2 and with low-frequency CBL, CSF3R, JAK2, and ETNKT. Some patients may have atypical features and other related disorders, such as atypical CML or chronic myelomonocytic leukemia, which generally have a poorer prognosis and do not respond to tyrosine kinase inhibitors (TKIs) as effectively.
WHO diagnostic criteria atypical chronic myeloid leukemia, BCR-ABL1 negative
| ■ Peripheral blood leukocytosis due to increased numbers of neutrophils and their precursors (promyelocytes, myelocytes,
metamyelocytes) comprising ≥10% of leukocytes |
| ■ Dysgranulopoiesis, which may include abnormal chromatin clumping |
| ■ No or minimal absolute basophilia; basophils usually <2% of leukocytes |
| ■ No or minimal absolute monocytosis; monocytes <10% of leukocytes |
| ■ Hypercellular bone marrow with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia in the erythroid and megakaryocytic lineages |
| ■ <20% blasts in the blood and bone marrow |
| ■ No evidence of PDGFRA, PDGFRB, or FGFR1 rearrangement, or PCM1-JAK2 |
| ■ Not meeting WHO criteria for BCR-ABL1+ CML, PMF, PV, or ET* |
Frontline treatments for CML include imatinib, dasatinib, bosutinib, and nilotinib. Imatinib has demonstrated favorable long-term outcomes, with high rates of cytogenetic and molecular remission and a 10-year survival rate of approximately 85%. However, resistance to imatinib can occur, and second-generation TKIs may be required. For advanced or refractory cases, allogeneic stem cell transplantation remains a viable option, offering a potential cure for about 70% of patients in chronic phase.
Reference
https://imagebank.hematology.org/image/61888 /chronic-myeloid-leukemia-presentation-in-peripheral-blood-1
 Dr. Madhumitha R M
Dr. Madhumitha R M
DNB General Medicine Resident
Kauvery Hospital, Chennai
 Dr. Sivaram Kannan
Dr. Sivaram Kannan
Clinical Lead & Chief Consultant Physician
Kauvery Hospital, Chennai
 Dr. Arshad Raja
Dr. Arshad Raja
Associate Consultant – Haematology and Haemato Oncology
Kauvery Hospital, Chennai