Advances in cardiac amyloidosis treatment: A review on Tafamidis
N. Dharsshini
Clinical Pharmacist, Kauvery Hospital, Trichy
Abstract
Cardiac amyloidosis poses diagnostic and therapeutic challenges. Tafamidis, a transthyretin stabilizer, shows promise in halting disease progression. This review examines its mechanism, efficacy, and safety. Clinical trials demonstrate reduced mortality and hospitalizations with tafamidis. Considerations include dosing, adverse effects, and cost-effectiveness. Early diagnosis is crucial for optimal management. Tafamidis offers a disease-modifying approach in cardiac amyloidosis. Integration into clinical practice requires careful consideration of patient characteristics. This review aims to inform clinicians about tafamidis’ role in improving outcomes. Optimizing care for cardiac amyloidosis patients remains a priority in clinical practice.
Introduction
Amyloidosis cardiomyopathy refers to cardiac involvement in amyloidosis, a group of disorders characterized by the extracellular deposition of abnormal protein fibrils in various organs and tissues, including the heart. In amyloidosis cardiomyopathy, the heart muscle becomes infiltrated by amyloid fibrils, leading to structural and functional abnormalities that can impair cardiac function. There are several types of amyloidosis, but the two most common types that can affect the heart are:
AL (light chain) amyloidosis: Caused by the deposition of abnormal immunoglobulin light chains.
ATTR (transthyretin) amyloidosis: Caused by the deposition of abnormal transthyretin protein, either due to a hereditary mutation (hereditary ATTR amyloidosis) or the wild-type protein (wild-type ATTR amyloidosis).
Patients with amyloidosis cardiomyopathy may present with symptoms of heart failure, such as shortness of breath, fatigue, leg swelling, and exercise intolerance. Other common symptoms may include palpitations, chest pain, and syncope (fainting). Some patients may also develop arrhythmias, including atrial fibrillation and heart block.
In amyloidosis cardiomyopathy, the abnormal protein fibrils deposit in the heart tissue, leading to disruption of the normal cardiac architecture. The accumulation of amyloid fibrils can cause thickening and stiffening of the heart muscle, impairing its ability to pump blood effectively. Additionally, amyloid deposition can disrupt the electrical conduction system of the heart, leading to arrhythmias and conduction abnormalities.[1]
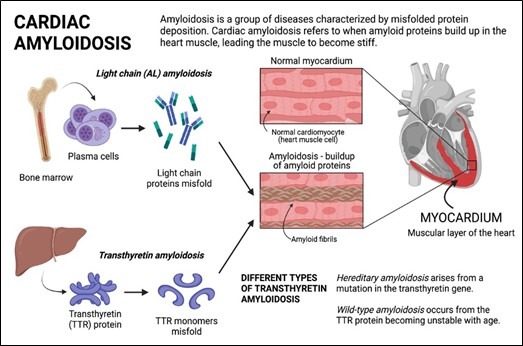
Fig (1): Pathogenesis of Cardiac Amyloidosis.1
A high level of clinical suspicion is needed to diagnose CA. In the past, the overemphasis on the presence of multi-organ involvement led to the diagnosis only being considered in cases with extreme extra-cardiac findings, such as macroglossia and periorbital purpura, which, although specific, are absent in ATTR-CA and present in only a minority of AL cases. When a patient has restrictive cardiomyopathy or left ventricular hypertrophy that is otherwise unexplained, a number of diagnostic red flags (Figure 1) can raise the right suspicion. Some helpful hints include a history of carpal tunnel syndrome in ATTR (especially if bilateral in a man), an atraumatic rupture of the biceps tendon, an inexplicable neuropathic pain, orthostatic hypotension, and a diagnosis of “hypertrophic cardiomyopathy” during the sixth decade of life. [2].

Fig (2): Factors to consider in Cardiac Amyloidosis
Treatment for Cardiac Amyloidosis

New Innovation—Tafamidis
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive, fatal disease caused by the deposition of transthyretin (TTR) amyloid fibrils in the myocardium, leading to cardiomyopathy and symptoms of heart failure
Tafamidis is a TTR kinetic stabilizer that inhibits tetramer dissociation, the rate-limiting step in TTR amyloid genesis. In the randomized Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT), tafamidis was shown to significantly reduce mortality and cardiovascular-related hospitalizations and to reduce decline in functional capacity and quality of life compared with placebo. Despite a recognized clinical benefit of tafamidis, its effect on cardiac function as measured by echocardiographic parameters has not been fully characterized. Three recent reports from single-center studies of tafamidis reported delays in cardiac structural and functional deterioration in patients with ATTR-CM treated with tafamidis compared with treatment-naive patients, thereby suggesting that tafamidis may attenuate the decline of cardiac function. [3-6]
Tafamidis is a transthyretin stabilizer. It works by binding to transthyretin, a protein produced by the liver that transports thyroid hormone and vitamin A in the blood. In ATTR cardiomyopathy, mutant forms of transthyretin misfold and aggregate, leading to the formation of amyloid fibrils that deposit in the heart tissue, causing damage. Tafamidis stabilizes the tetrameric structure of transthyretin, preventing it from misfolding and forming amyloid fibrils. By stabilizing transthyretin, tafamidis slows down the progression of ATTR cardiomyopathy and helps preserve heart function.
Tafamidis is typically admindistered orally in the form of tablets. The recommended dosage of tafamidis for ATTR cardiomyopathy is usually 80 mg once daily for adults.
Tafamidis is generally well-tolerated, with most adverse effects being mild to moderate in severity.
Common side effects may include gastrointestinal symptoms (such as diarrhea, abdominal pain, and nausea) and urinary tract infections.
According to The Lancet 2023, Wang, Jie et al., Patients with ATTR-CM, tafamidis treatment was associated with a low risk of all-cause death, heart transplant, heart assist device implantation, heart failure exacerbations or hospitalizations. In addition, tafamidis treatment may decrease deterioration in LVEF in patients with wtATTR. Further research, encompassing larger sample sizes and long-term follow-up, is warranted to evaluate the efficacy of tafamidis in the treatment of ATTR.[7]
Tafamidis represents a significant advancement in the treatment of ATTR cardiomyopathy by targeting the underlying disease process of transthyretin misfolding and aggregation. It provides an important therapeutic option for patients with this rare and debilitating condition. However, as with any medication, its use should be guided by clinical judgment and individual patient characteristics, and patients should receive regular monitoring and follow-up cares
Reference
- Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation.2017;135(14):1357-1377. doi:10.1161/CIRCULATIONAHA.116.024438.
- https://ucsfhealthcardiology.ucsf.edu/care/clinical/amyloid
- Maurer MS, Schwartz JH, Gundapaneni B, et al; ATTR-ACT Study Investigators. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007-1016. doi:1056/NEJMoa1805689PubMedGoogle ScholarCrossref.
- Rettl R, Mann C, Duca F, et al. Tafamidis treatment delays structural and functional changes of the left ventricle in patients with transthyretin amyloid cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2022;23(6):767-780. doi:1093/ehjci/jeab226PubMedGoogle ScholarCrossref
- Rettl R, Duca F, Binder C, et al. Impact of tafamidis on myocardial strain in transthyretin amyloid cardiomyopathy. Amyloid. 2023;30(1):127-137. doi:1080/13506129.2022.2131385PubMedGoogle ScholarCrossref
- Giblin GT, Cuddy SAM, González-López E, et al. Effect of tafamidis on global longitudinal strain and myocardial work in transthyretin cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2022;23(8):1029-1039. doi:1093/ehjci/jeac049PubMedGoogle ScholarCrossref
- Tafamidis treatment in patients with transthyretin amyloid cardiomyopathy: a systematic review and meta-analysis Wang, Jie et al. eClinicalMedicine, Volume 63, 102172
