Understanding Thalassemia: A comprehensive overview
Vinitha. M*
Group Clinical Pharmacist, Kauvery Hospital, Trichy
Definition
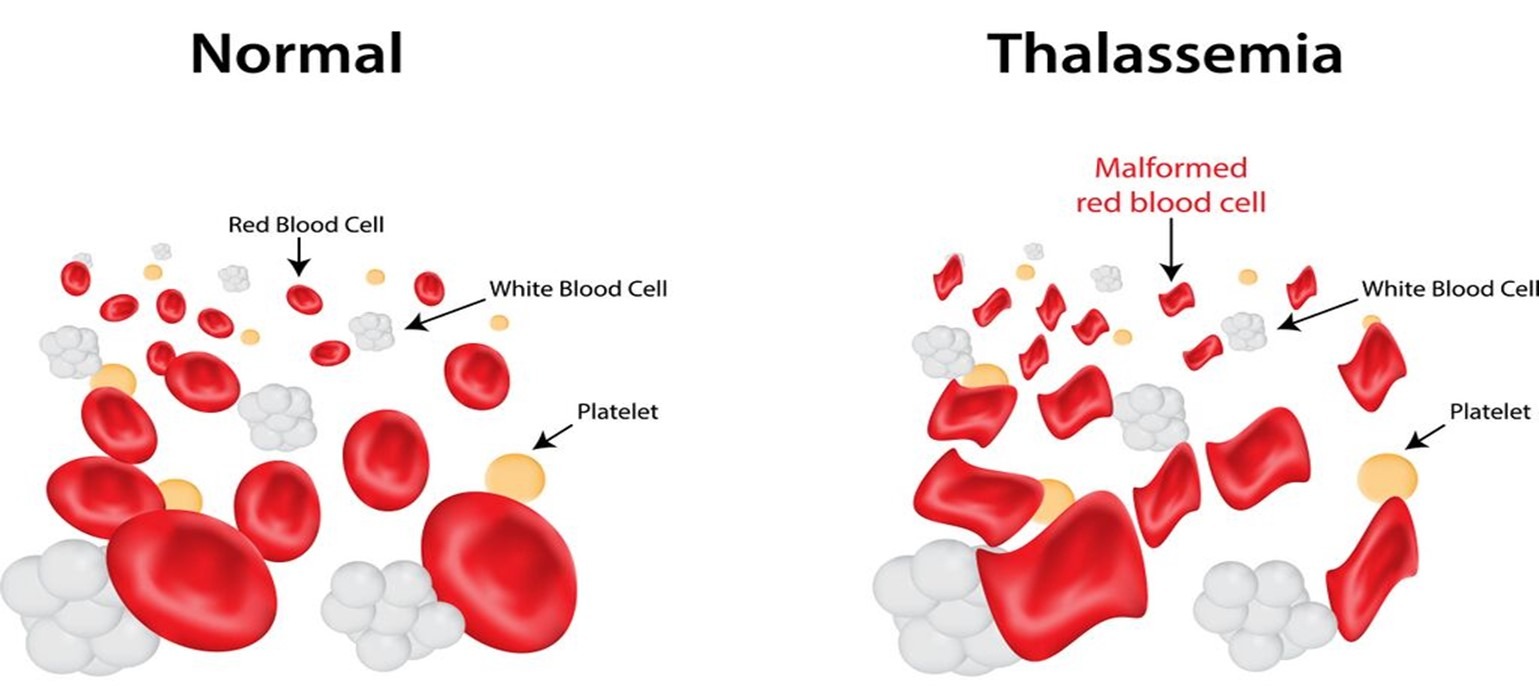
Thalassemia is a group of inherited blood disorders characterized by reduced hemoglobin production. Hemoglobin is the protein in red blood cells that carries oxygen throughout the body. Thalassemia results in anemia, which can vary in severity.
Thalassemia is an inherited blood disorder that affects hemoglobin production. This condition results in anemia, which can range from mild to severe. Understanding thalassemia, its types, symptoms, diagnosis, and treatment options is crucial for managing the condition effectively and improving the quality of life for those affected.
Types of Thalassemia
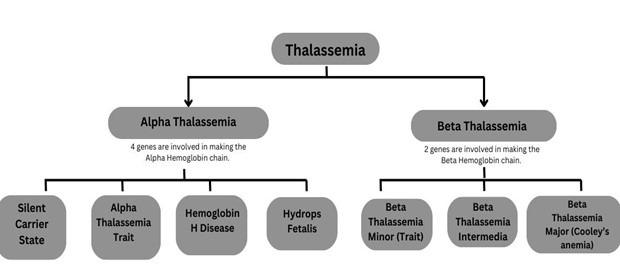
Alpha Thalassemia
It is caused by mutations in the HBA1 and HBA2 genes, which encode the alpha-globin subunits of hemoglobin. There are four types, depending on how many of the four alpha-globin genes are affected:
- Silent Carrier State: One gene is affected. Individuals are typically asymptomatic.
- Alpha Thalassemia Trait: Two genes are affected. Individuals may have mild anemia.
- Hemoglobin H Disease: Three genes are affected. Symptoms include moderate to severe anemia, splenomegaly, and bone deformities.
- Hydrops Fetalis: All four genes are affected. This condition is usually fatal before or shortly after birth.
Beta Thalassemia
It is Caused by mutations in the HBB gene, which encodes the beta-globin subunits of hemoglobin. There are three types based on the severity of the mutations:
- Beta Thalassemia Minor (Trait): One gene is affected. Individuals are typically asymptomatic or have mild anemia.
- Beta Thalassemia Intermedia: Both genes are affected, but the mutations allow for some beta-globin production. Symptoms range from moderate to severe anemia.
- Beta Thalassemia Major (Cooley’s Anemia): Both genes are affected, leading to little or no beta-globin production. Symptoms are severe and include severe anemia, growth retardation, and skeletal abnormalities.
Flowchart: Types of Thalassemia
Medical history and physical examination
- Family history of anemia or known thalassemia.
- Symptoms such as fatigue, weakness, pallor, jaundice, and growth issues.
- Physical examination may reveal pallor, jaundice, splenomegaly, and skeletal deformities.
Laboratory Tests
- Complete Blood Count (CBC): Shows anemia, reduced mean corpuscular volume (MCV), and red blood cell (RBC) count abnormalities.
- Hemoglobin Electrophoresis: Identifies abnormal hemoglobin types and quantifies hemoglobin A2 and F.
- Genetic Testing: Confirms mutations in the HBA or HBB genes.
Imaging and Other Tests
- Ultrasound: Assesses spleen and liver size.
- Bone Marrow Examination: In some cases, to evaluate erythropoiesis.
Treatment
Mild Thalassemia
- Often requires no treatment.
- Regular monitoring and supportive care, such as folic acid supplements.
Moderate to Severe Thalassemia
- Regular Blood Transfusions: To maintain adequate hemoglobin levels and manage anemia.
- Iron Chelation Therapy: To prevent iron overload due to frequent transfusions. Excess iron can be toxic and lead to organ damage. There are several types of iron chelation therapy, which involve different chelating agents that bind to iron and help remove it from the body. Medications include Deferoxamine, Deferasirox, and Deferiprone.
- Folic Acid Supplements: To support red blood cell production.
- Splenectomy: May be considered in cases of severe splenomegaly.
- Bone Marrow or Stem Cell Transplant: The only potential cure, usually recommended for severe cases, especially in children.
Experimental and Future Treatments
- Gene Therapy: Research is ongoing to correct the genetic mutations causing Thalassemia.
- New Medications: Such as Luspatercept, which is designed to improve hemoglobin levels.
The latest advancements in thalassemia treatment
Gene Therapy
- Gene therapy approaches using lentiviral vectors, gene editing techniques like CRISPR-Cas9, and induced pluripotent stem cells are showing promising results for treating beta-thalassemia . These therapies aim to correct the genetic defect causing thalassemia.
- One gene therapy product, betibeglogene autotemcel (beti-cel), has reached phase 3 clinical trials with promising results in reducing transfusion requirements for patients with transfusion-dependent beta-thalassemia.
Other Emerging Treatments
- Drugs like Luspatercept and Roxadustat can help stimulate red blood cell production and reduce transfusion needs in beta-thalassemia patients.
- Hematopoietic stem cell transplantation, though limited by donor availability and complications, remains a potential curative option for Thalassemia.
- Approaches to reactivate fetal hemoglobin production, such as with hydroxyurea, can also help ameliorate symptoms in some Thalassemia patients.
Clinical Data on Thalassemia in Kauvery hospital, Cantonment
Table 1. Age wise Distribution of patients diagnosed as Thalassemia
| Age range | Number of patients | Percentage of patients |
|---|---|---|
| <1 Year | 8 | 3% |
| 1 - 5 Year | 99 | 42% |
| 6 - 10 Year | 63 | 27% |
| 11 - 15 Year | 48 | 21% |
| 16 - 20 Year | 14 | 6% |
| 21 - 25 Year | 2 | 1% |

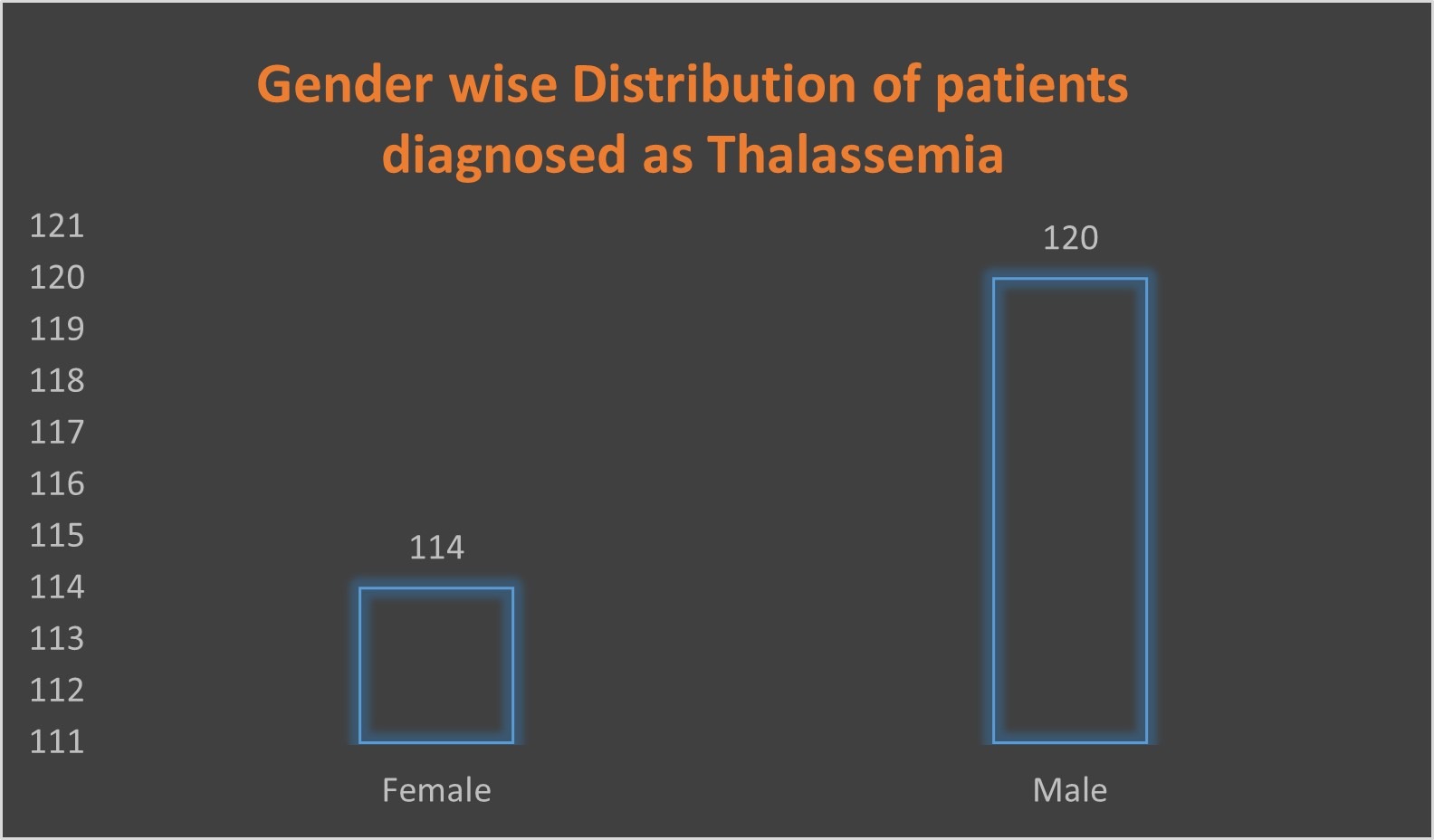
Table 2. Gender wise Distribution of patients diagnosed as Thalassemia
| Gender Wise | Number of patients | Percentage of patients |
|---|---|---|
| Female | 114 | 49% |
| Male | 120 | 51% |
Discussion
Though it was a deadly disease, we can cure that with the emerging techniques
From January to June 2024 we retrospectively collected and analyzed a data of 234 patients from the Kauvery hospital cantonment diagnosed as thalassemia, which showed more male patients (51%) when compared with female patients (49%).
In that study population, 42% patients were between the age group of 1-5 years followed by 27% of patients on 6-7 years age group.
From the results of study conducted by Dr. Vinoth et al in kauvery hospital also confirms that the most vulnerable age group is 1-5 years (40%) followed by 6–10 years (27%). Out of this population, 12 patients were managed with single chelation therapy and 15 patients were managed by double chelation therapy.
Conclusion
Overall, the treatment landscape for Thalassemia is rapidly evolving, with gene therapies and other novel agents providing new hope for reducing transfusion requirements and managing complications like iron overload for these patients. Iron chelation therapy is crucial in managing iron overload and preventing its complications. The choice of chelating agent and the treatment plan should be tailored to each patient’s individual needs and medical conditions.
Acknowledgement
Dr. Vinod Gunasekaran
His paper on “Clinico Epidemiological profile of children with Thalassemia Major in Kauvery Hospital, Trichy, is being published in KAUVERIAN on 08 Aug 24.
Nightingale Journals
- Message from Dr. Manivannan Selvaraj, Founder and Managing Director, Kauvery Hospitals
- EDITORIAL
- Editorial Board
- Voices from the field!
- Junior nurses in Kauvery Hospital on the frontline against the COVID-19 pandemic
- Challenges in nursing care in achieving a successful outcome to a Liver Transplant on a 2-year old child with life threatening genetic condition
- Nursing management of a multi-organ transplant (Liver and Kidney) on an adolescent
- Successful discharge of a four year old baby with life threatening gas gangrene and compartment syndrome
- The revolutionary effect of a nursing-driven initiative to reduce ICU re-admissions
- Nephrotic syndrome: A case report
- Nursing Care of Patient with Thoracic Endo vascular Aortic Repair (TEVAR)
- Efficient Nursing Care equals Early Recovery
- Umbilical Cord Blood Banking is a lifesaver for your family, “Create one life, save another”: A review
- Challenges associated with wearing gloves
- Certificate Course on Infection Control 2022
- The ANEI Young Leader Award, 2022, comes to Kauvery Hospitals!
- The AHPI Nursing Excellence Award 2022, comes to Kauvery Hospital, Tennur
- Editorial
- Editorial Board
- Caring is the Essence of Nursing
- To be patient and calm! Why I Love Working as an Emergency Room Nurse
- Infective Endocarditis
- Infective Endocarditis
- Patent Ductus Arteriosus
- HOSPITalk 2023 – Clinical Governance
- Cardiac healthy diet
- Healthy diet for people working night shifts
- Chimeric antigen receptor (car) T cell therapy
- Human papillomavirus (HPV) vaccine
- Kauvery Hospital Journey on Winning National Level CII QC Competition Against the Industrial Sectors
- செவிலியர்கள் தின வாழ்த்து கவிதை
- அன்பு காவேரி
- Hepatology for nurses
- Role of Physiotherapy in Traumatic Brain Injury: A case report
- My experience at Kauvery Hospital, Salem
- Editorial
- Editorial Board
- Benign Paroxysmal Positional Vertigo (BPPV): A Case Series
- Nutrition for Nurses
- Cardiovascular effects of sodium – glucose co-transporter- 2 inhibitor, Glucagon-like Peptide-1 Receptor Agonists and Dipeptidyl-peptidase 4 inhibitors
- Brief Guide on Food Safety Standards to be Adopted Summer in South India
- Kauvery Hospital: Pioneering the Future of Healthcare with IoT, Wins Healthcare Asia Awards.
- மருத்துவர்கள் தின வாழ்த்து கவிதை
- காவேரியனின் தன்னம்பிக்கை
- வெற்றியின் பாதை
- Editorial
- Editorial Board
- A six-year-old girl gets a Permanent Pacemaker implanted!
- Cardio Renal Amyloidosis (Non-Secretory Myeloma)
- Deep Vein Thrombosis, treated with Angiojet Percutaneous Mechanical Thrombectomy (PMT): A Case Report
- Emergency CABG in Acute MI
- Meatotomy
- Septic Shock with Multiple Organ Dysfunction Syndrome: A Case Report
- Stab Injury: A Case Report
- The Journey of Dialysis Unit at Hosur
- The measure of life is not its duration but its donation
- Innovation at the frontline of Nursing: A Review
- A case study on hydatid cyst
- Untold History of British Scientist: Contributed to the Discovery of DNA
- Neurology for Nurses
- Poem
- மருத்துவாலயம்
- EDITORIAL
- Editorial Board
- Biological or circadian clock
- A novel direct thrombin inhibitor from tick salivary transcriptoms: A review
- Nursing Care of a Patient with Coarctation of aorta (COA)
- Nursing care of patient with aortic dissection
- Wireless moonlight: An ultra-short case study
- Acute liver failure: A case report
- We nurses hand-hold a patient on a long walk down a dark COVID road
- Effectiveness of segmental breathing and active cycle of breathing technique in the management of dyspnoea among covid-19 patients
- EDITORIAL
- Editorial Board
- I am a proud to be a Liver Transplant Nurse!
- Head Injury: A case report
- Postpartum/Peripartum Cardiomyopathy
- Nursing Care of Patient with Myocardial Infarction
- Nursing challenges faced in care of patient with Blunt Injury Abdomen
- Salicylate poisoning
- Vascular Access Management (VAM) training program for nurses
- Therapeutic Nutrition among Critical care patients
- Diagnostic Image
- EDITORIAL
- Editorial Board
- Guideline-directed drug treatment for heart failure
- Dilated cardiomyopathy
- IV Proton Pump Inhibitors (PPIs): What are the true indications?
- Role of physiotherapy in GBS patients during hospitalization:A case presentation
- Pulmonary Thromboembolism
- Paraquat poisoning, an emerging problem, a challenging outcome
- Achievements of today are the stepping stones for success tomorrow!
- Diagnostic Image
- Chronic obstructive pulmonary disease: A case report
- EDITORIAL
- Editorial Board
- The Good Nurse
- Success story of a patient with Haemangioma
- Role of physiotherapy in Bell’s Palsy at outpatient department
- Arrhythmogenic Right Ventricular Dysplasia: A Cardiomyopathy
- Systemic Lupus Erythematosus (SLE)
- Teamwork makes a Dreamwork
- Cholecysto Cutabeous Fistula, an emerging problem: a challenging outcome
- Adhesive small bowel obstruction: Nutrition care process
- Diagnostic Image
- Editorial
- Editorial Board
- Quality improvement project to reduce the incidence of ventilator-associated pneumonia
- Rare disease, desired outcome
- Management of patients with acute myocardial infarction with ischemic stroke
- Left mediastinal tumor excision
- Esophageal varices grade III: A case report
- TB meningitis: A case report
- Nutrition care process for Sigmoid Diverticulitis
- Nutritional management of patient who underwent emergency laparotomy and GIST
- Nutritional management of gestational diabetes mellitus
- Nutrition and drug interaction
- மக்களின் நம்பிக்கை!
- காவேரித்தாய் – 2
- ஊசியின் மகத்துவம்
- Editorial
- Editorial Board
- Resilient and Empowered: The Unstoppable Force of Women’s Spirit
- “Global Perspectives on Patient Safety: A Recap of the International Patient Safety Conference – 13th to 14th Feb 2023”
- Acute Pancreatitis: the nutrition care process
- Role of diet in mitral valve replacement: A case presentation
- Our colleague’s SVT (Supra Ventricular Tachycardia)!
- Acute respiratory distress syndrome: A case report
- To prevent the negative impact of Inj. Amphotericin among Mucormycosis patients
- Diagnostic Image
- Effective Adherence of Checklist through Digitalisation
- Nephrology for Nurses
- “உயிரைக்காக்கும் உயரியதானம்”
- மக்களின் நம்பிக்கை
- Editorial
- Editorial Board
- SVT (Supra Ventricular Tachycardia)
- Tender loving care of patient with DVT after IVF (in vitro fertilization)
- ASD Surgical Closure
- Quality improvement project to reduce the risk of cross infection from wall mounted suction apparatus
- Quality Improvement Project on Crash Cart Management with numbered seal
- Log the Indian Millets
- Crystal your body by dint of seven Crystal Seeds!
- Role of Glucagon
- Educative Image: Department of Orthopaedics
- Nephrology for Nurses! – Part 2
- EDITORIAL
- Editorial Board
- இதயநகரத்தின் செவிலியர்கள் தின வாழ்த்து
- My experience as a patient
- A study on Anidulafungin therapy in oncology patients in a tertiary care hospitals
- A case study on Heart Transplantation
- Your Physiotherapist’s talks! Know your Moves
- Impact of air pollution on human health
- 4th QOK Group Level Competition 2023
- 7th International conference of CAHO
- செவிலியர் எனும் தாய்
- HEPATOLOGY FOR NURSES
- Editorial
- Editorial Board
- Unknown etiology, thoughtful management, gratifying outcomes!
- Antiphospholipid Syndrome(APLS) in a man
- World Organ Donation Day Celebration at Kauvery Hospital, Hosur
- Approach to cardiac arrest
- Cardiac Tissue Viability Study
- Dietary management after Mitral Valve Replacement
- Clipping of a Cerebral Aneurysm at Nellai
- Care of Chronic Suppurative Otitis Media (CSOM)
- Pituitary Macroadenoma: A case report
- Superior mesenteric artery thrombosis
- Pharmacists: The Silent Heroes of the Health Care System
- Educative Image
- Poem
- A battle to win a baby
- 5S Sustenance Level-Up for Salem, Hosur and ECB Units
- Editorial
- Prevention of accidental removal of tube and drains: A case report
- Clinical Report Workshop: An initiative by the Clinical Governance Team
- Burr Hole – evacuation of right frontoparietal chronic Sub Dural Hematoma and left parietal SDH
- Balloon Mitral Valvotomy
- Intra Pulmonary Thrombolysis
- Nephrotic Syndrome: A case report
- Trans catheter Aortic Valve Implantation (TAVI)
- Tetralogy of Fallot (TOF): Echocardiography
- Patent Ductus Arteriosus (PDA)
- A souvenir to remember the Nurses day 2024: Celebration of the Guardian angel of Healthcare
- Flying Cherub
- காவேரியின் செவிலியராய்!
- காவேரியின் பசுமை புரட்சி
- Editorial
- Care of patient with Interstitial Lung Disease
- Ovarian Torsion: A case report and discussion
- Ovarian torsion: A case report
- Acute Respiratory Distress Syndrome: A case report
- Shadow reports of heart city’s valve diseases and clinic
- Innovative strategy, empowerment and amendment of guidelines to prevent IV complications
- Effectiveness of Nurse- fabricated innovative device (K-Brace) on prevention of phlebitis
- Organ Donor Hero!
- Understanding Thalassemia: A comprehensive overview
- Pressure Injuries—the sensitive indicator
- The sixth QOK Competition April 2024
- Editorial
- Infective Endocarditis (IE)
- A case report on Bull Gore Injury
- Free Flap for traumatic raw area: A case report
- The Story of the “First Cry”
- Case series on drug-induced anaphylactic shock
- Clinical therapeutics: The adrenergic system and related drugs
- Flying angel experience
- From QOK to KOACH: A journey of continuous improvement
- Poem – காவேரியின் நவீன ஐந்து எஸ்(5S) மாடல்
- Editorial
- Editorial Board
- Attempted Hanging: A case series
- Sub Arachnoid Hemorrhage due to Cerebral Aneurysm
- Acute Pulmonary Thromboembolism with Systemic Lupus Erythematous
- Down Syndrome with Severe Pulmonary Stenosis
- Bentalls Procedure
- Glycogen Storage Disease (GSD)
- Nursing Care of Patient with Penetrating Chest Injury (Left Chest Wall)
- A child with Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP) – Guillian- Barre Syndrome (GBS)
- Periampulatory carcinoma (Whipple’s procedure)
- Assisted delivery with Forceps Extraction
- Thrilling Moment on a Successful transportation of the cadaver donor liver to its recipient
- Editorial
- Transcatheter Aortic Valve Implantation (TAVI)
- Cautery Burns: A Clinical Audit
- Automatic Implantable Cardioverter Defibrillator (AICD)
- Thymectomy
- Deep Vein Thrombosis: A case report
- Mesentric Neoplasm
- QT Syndrome
- Healthy diet, affordable for all – Fuel for the Future
- Personalizing 5-FU Treatment in Head and Neck Cancer: A TDM Pilot Study
- Rapid Review of CNE -Nursing Challenges in Coronary Artery Diseases
- Preconference Workshop on International patient safety Goals (IPSG)
- Editorial
- Cerebral malaria: Management with artesunate
- Hypertrophic cardiomyopathy: A case report
- Incredible challenges and outcomes: A case report
- Prevention of extravasation in Oncology unit
- Management of germ cell tumors: A review
- Monocytes: The mysterious cell on the CBC
- Efficacy of individualized use of a multisensory integrative environment on engagement: In children with sensory modulation disorder
- Dietary guidelines and food safety: For immuno-suppressed/compromised patients
- Nutritional management: For a patient with triple vessel disease
- இயன்முறை மருத்துவமும் மறுவாழ்வும்
- உயிர் காக்கும் தானம்
- மாற்றத்தை விதைக்கும் திறனாளிகள்
- EDITORIAL
- EDITORIAL BOARD
- CEREBRAL MALARIA: A CASE REPORT
- CEFEPIME – TAZOBACTUM – INDUCED FLUID – FILLED BLISTERS (BULLOUS LESIONS): A CASE REPORT
- BLOOD TRANSFUSION REACTIONS: AN OVERVIEW
- EFFECTIVENESS OF REHABILITATIVE APPROACH-BASED MANAGEMENT FOR CHILDREN WITH JAPANESE ENCEPHALITIS (JE)
- THE EFFECT OF NUTRITIONAL COMPOSITION ON THE GLYCEMIC INDEX AND GLYCEMIC LOAD VALUES
- KCHS PRATIDHI RISES TO THE CHALLENGE: A TRIUMPH AT THE NATIONAL QC CONVENTION
- JOURNAL SCAN FOR THE CLINICAL PHARMACIST
- காவேரித்தாய் – 4
- EDITORIAL
- INSTRUCTIONS TO AUTHORS
- AORTIC ANEURYSM WITH PARAVERTEBRAL COLLECTION
- DEEP VEIN THROMBOSIS AT RIGHT UPPER LIMB: A CASE REPORT
- ENTEROVIRUS ASSOCIATED MENINGOENCEPHALITIS: A CASE REPORT
- CONGENITAL HEART DISEASE (ASD – OS): A CASE REPORT
- FUNCTIONAL SHORT GUT SYNDROME: A CASE CAPSULE
- PERI-OPERATIVE CARE DURING CYTOREDUCTION SURGERY (CRS) WITH HYPERTHERMIC INTRAPERITONEAL CHEMOTHERAPY (HIPEC): KAUVERY EXPERIENCE.
- A CASE REPORT: MYELOPROLIFERATIVE NEOPLASMS (MPNS) WITH CABG
- OVERVIEW OF BREAST FEEDING: A REVIEW
- PITUITARY MACRO ADENOMA-TRANS-NASAL TRANS-SPHENOIDAL ENDOSCOPIC EXCISION
- JOURNAL SCAN: A CASE REVIEW OF IMMEDIATE CLINICAL SIGNIFICANCE, HARVESTED FROM MAJOR INTERNATIONAL JOURNAL
- தயக்கம் தவிர்
- Editorial
- Editorial Board
- Case Report: A success story of IVUS: In a PTCA
- Case Report on Ovarian Torsion
- Nursing care for a child with Thrombophlebitis
- Drug Induced Hypersensitivity to Salazopyrin: A case report
- The Phytonutrients — 365.25 Days/52 Weeks of Phytonutrients help’s to develop our shape
- Role of diet in coronary artery disease
- 5S Cross Unit Assessment 2024
- என்னுள் 5S
- பெண்ணே பெருமை கொள்
- Journal scan
- Editorial
- Instructions for Authors
- Ventricular Septal Defect (VSD): Echocardiography
- Steven Johnson Syndrome: A case report
- A case review on Capsule Endoscopy
- Savoir Faire: A management for mass causality
- Case report on Gouty Arthritis
- Management of a road traffic accident victim with severe head injury, and aspiration: A case report
- A case report on inhalation of chlorine gas
- ST segment elevation during Treadmill exercise test in a patient without prior Myocardial Infarction
- History and evolution of Corporate and Clinical governance
- The need of Human Papilloma Virus (HPV) vaccine: A review
- Futuristic face of resuscitative Centhaquine for hypovolemic shock: A Review
- Boost your health in this summer
- A data-driven approach to patient care: Kauvery Hospital pioneers six sigma in healthcare
- Fascinating experience as a flying angel
- விளையாட்டுத் திடல்
- எங்கள் காவேரி
- Editorial
- Triple Bypass: A case report
- Boerhaave Syndrome with Mediastinitis
- Right sided Infective Endocarditis
- Marburg virus disease: A systematic review
- Advances in cardiac amyloidosis treatment: A review on Tafamidis
- Role of salt in human health
- Learning by playing: 5S makes school a breeze
- நம் காவேரி
- செவிலிய தேவதைகள்
- Editorial
- A case report: Melioidosis
- Type IV-A Choledochal cyst
- Nursing care of the patient with right lower lung foreign body and Bronchiectasis: Treated surgically with lobectomy
- Care of patient with OPC poisoning
- The Rosai-Dorfman disease presents with extranodal involvement and diagnostic challenges
- Open stab injury by a Bull
- In-house Continuing Nursing Education (CNE) on mastering the complexities of critical illnesses at Kauvery Hospital, Tennur, 2024
- Effectiveness of trigger point release and ultrasound therapy on Trapezitis
- Kauvery Hospital Trichy region’s journey towards 5S model hospital recognition
- A Triumphant journey to 5S sustenance level I
- Poem – இரத்த தானம்
- Poem – ஆசிரியர் தின நல்வாழ்த்துக்கள்
- Poem – ஆசையான ஆசான்
- Poem – அவள் ஓர் அறிவியல்
- Editorial
- When Banding Breaks, New Paths Awaken: The BRTO Revelation
- Smile Therapy
- Multidisciplinary approach to Thermal Burns
- Deep Brain Stimulation for Parkinson’s disease: A case report
- Zieve’s Syndrome: A review
- Acute Pulmonary Thromboembolism
- MPI scan guided revascularization in acute anterior wall Myocardial Infarction
- Ketogenic diet for Epilepsy: A case report and review
- Dietary management: Carcinoma in left buccal mucosa
- Malignant Middle Cerebral Artery (MCA) infarct and surgical decompression: Pre-op and post-op CT brain findings
- Cleistanthus collinus (Oduvanthalai poisoning): A case report
- My Experience as a Flying Angel
- In-house Continuing Nursing Education (CNE) on “Rapid Response Mastery
- Kauvery Hospital Salem’s Journey of 1st Ever Model Hospital
- மனமும் வெற்றியின் ரகசியமும்
- Editorial
- Against all odds: A road accident survivor’s journey to healing at Kauvery Hospital
- Clinical Case Report: Managing Hansen’s Disease in a 20-Years young girl
- Bilateral Internal Thoracic Artery Grafting for CABG
- Intra Pulmonary Thrombolysis
- A Case Report on Methotrexate-Induced Pancytopenia
- An Adult with an Atrial Septal Defect Presenting with a Brain Abscess
- Typhoid, a Prospective Observational Study
- Vancomycin – Therapeutic Drug Monitoring
- Cardiac’s Myxoma
- Mitral valve replacement
- Harmful effects of preservatives (Class 1) on Food Items
- In house Continuing Nursing Education (CNE) on “Shaping Excellence in Critical Care Nursing.” At Kauvery hospital, Cantonment.
- Poem – செவிலியர்
- Poem – ஒருபோதும் கேட்காதீர்கள்: “உனக்கு என்ன வேண்டும் என்று”
- Editorial
- A case report on Carbuncle
- Reverse Shoulder Arthroplasty: A case report
- A case report on severe dental caries with advanced lesions
- Supra ventricular Tachycardia: A case report
- A case of pernicious anaemia due to vitamin B12 deficiency
- A Journey of Miracles: Life Beyond the Deadly Trials for My Father
- A Victory day for CNE
- A Sapient Voyage – QCFI
- Tracheostomy: An overview
- முன்கூட்டியே கண்டறிவோம் புற்றுநோயை
- Editorial
- Emergency CABG for young female patient with critical coronary artery disease
- Meningomyelocoele: A case report and discussion
- Case study on Multiple Cranial Nerve Palsy and Necrotizing Pneumonia: The physiotherapy management
- Role of Physiotherapy in ACL Rehabilitation: A case report
- ASD Device Closure: Case report and discussion
- In-House-Continuing Nursing Education (CNE) on “Effective Nursing Strategies for Renal Transplantation” at Kauvery Hospital, Tennur
- காவேரியின் வாக்ஹோலிக் நடைபயிற்சி
- புத்தாண்டு
- Editorial
- Artificial Intelligence in Nursing: Enhancing Care and Reducing Burnout
- Report on comprehensive wound care workshop—elevating nursing excellence at Kauvery Hospital
- Cerebellopontine angle tumor
- Patient acuity score: Staffing plan
- Acute Respiratory Distress Syndrome
- Coronary Artery Disease and Carotid Stenosis: A dual threat
- Early-onset diabetic foot ulcers in CKD
- Nursing case study report: Reconstructive surgery for congenital TMJ ankylosis
- Care of severe ARDS and H1N1 Positive
- Whipple Procedure: A case report
- A milestone to remember in my career
- Poem – காதல்
- Poem – ஆரோக்கிய வாழ்வு – 2
- Editorial
- Management of Myelodysplastic Syndrome (MDS) with Probable Fungal Pneumonia
- Thrombotic Microangiopathy and Renal Cortical Necrosis in a Postpartum Patient: A rare and complex presentation
- Rising Star in Health care
- Systemic Lupus Erythematosus: A case report and discussion
- Effectiveness of Cardiopulmonary Resuscitation( CPR) and its Outcome
- Guillain-Barre syndrome
- Radiation-free ERCP in pregnancy
- Utilization of injection Sovateltide for acute ischemic stroke
- A case of severe malaria complicated by concurrent H 3 N 2 influenza infection: Diagnostic and therapeutic challenges
- Pulmonary Function Test Concepts
- Rapid Review of CNE – Enhancing Nursing Practice in Arrhythmia Management: Evidence Based Strategies
- நூறைக் கடந்த காவேரியின் மருத்துவ இதழ்(ஜர்னல்)
- பெண் என்பவள்
- வியக்கத்தகும் அதிசயமே! கண்டு வியக்கிறேன்
