Journal scan: A review of 10 recent papers of immediate clinical significance, harvested from major international journals
From the desk of the Editor-in-Chief
(1). Doki Y. Nivolumab Combination Therapy in Advanced Esophageal Squamous-Cell Carcinoma. N Engl J Med. 2022;386:449-62.
Background
First-line chemotherapy for advanced esophageal squamous-cell carcinoma results in poor outcomes. The monoclonal antibody nivolumab has shown an overall survival benefit over chemotherapy in previously treated patients with advanced esophageal squamous-cell carcinoma.
Methods
In this open-label, phase 3 trial, we randomly assigned adults with previously untreated, unresectable advanced, recurrent, or metastatic esophageal squamous-cell carcinoma in a 1:1:1 ratio to receive nivolumab plus chemotherapy, nivolumab plus the monoclonal antibody ipilimumab, or chemotherapy. The primary end points were overall survival and progression-free survival, as determined by blinded independent central review. Hierarchical testing was performed first in patients with tumor-cell programmed death ligand 1 (PD-L1) expression of 1% or greater and then in the overall population (all randomly assigned patients).
Results
A total of 970 patients underwent randomization. At a 13-month minimum follow-up, overall survival was significantly longer with nivolumab plus chemotherapy than with chemotherapy alone, both among patients with tumor-cell PD-L1 expression of 1% or greater (median, 15.4 vs. 9.1 months; hazard ratio, 0.54; 99.5% confidence interval [CI], 0.37 to 0.80; P<0.001) and in the overall population (median, 13.2 vs. 10.7 months; hazard ratio, 0.74; 99.1% CI, 0.58 to 0.96; P=0.002). Overall survival was also significantly longer with nivolumab plus ipilimumab than with chemotherapy among patients with tumor-cell PD-L1 expression of 1% or greater (median, 13.7 vs. 9.1 months; hazard ratio, 0.64; 98.6% CI, 0.46 to 0.90; P=0.001) and in the overall population (median, 12.7 vs. 10.7 months; hazard ratio, 0.78; 98.2% CI, 0.62 to 0.98; P=0.01). Among patients with tumor-cell PD-L1 expression of 1% or greater, a significant progression-free survival benefit was also seen with nivolumab plus chemotherapy over chemotherapy alone (hazard ratio for disease progression or death, 0.65; 98.5% CI, 0.46 to 0.92; P=0.002) but not with nivolumab plus ipilimumab as compared with chemotherapy. The incidence of treatment-related adverse events of grade 3 or 4 was 47% with nivolumab plus chemotherapy, 32% with nivolumab plus ipilimumab, and 36% with chemotherapy alone.
Conclusions
Both first-line treatment with nivolumab plus chemotherapy and first-line treatment with nivolumab plus ipilimumab resulted in significantly longer overall survival than chemotherapy alone in patients with advanced esophageal squamous-cell carcinoma, with no new safety signals identified
(2). Theodoropoulos KC. Under-sensing by a temporary pacemaker after cardiac surgery and ventricular fibrillation. The Lancet 2022;399(10325):605-94.
An 80-year-old man with a 6-month-history of shortness of breath on minimal exertion and fatigue was admitted to our unit for a planned operation to replace his stenotic aortic valve. The surgery was uneventful and a temporary ventricular epicardial pacing lead was placed on the free wall of the right ventricle. The patient remained haemodynamically stable in sinus rhythm post-surgery and was extubated 8 h later. The following day he was transferred from intensive care to our high dependency unit with the temporary epicardial pacemaker in VVI mode: stimulation amplitude 10 V, sensitivity 1 mV, and back-up pacing rate of 44 beats per min. A pacing check at that time showed no problems.
2 days after the operation the patient had a cardiac arrest during mobilisation and physiotherapy; chest compressions were started, and the patient was re-intubated. The defibrillator showed ventricular fibrillation and he was shocked into sinus rhythm using 200 joules biphasic.
A telemetry review showed a spike on a T wave as a possible cause of the cardiac arrest: sinus rhythm was present during the first two cardiac cycles-heart rate 76 beats per min-followed by a premature pacing spike which produced a paced cardiac cycle. Another intrinsic cardiac cycle then followed. However, during the repolarisation phase of this cycle, a second pacing spike was seen falling on the T wave; this spike-triggered polymorphic ventricular tachycardia that apparently degenerated into ventricular fibrillation.
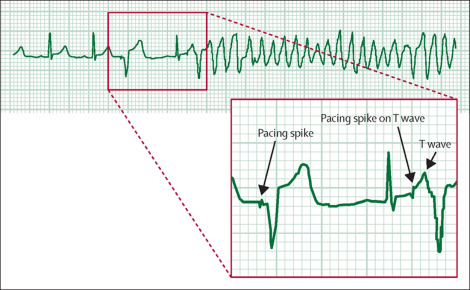
The patient was stabilised post-resuscitation and extubated 4 h later; he recovered well and was allowed home 12 days later. 1 month later he was well and asymptomatic when reviewed in our outpatient clinic.
Indications for temporary cardiac pacing include the management of bradyarrhythmias-specifically atrioventricular block-and both atrial and ventricular tachyarrhythmias. In cardiac surgery, insertion of temporary epicardial pacing wires is common practice; it both optimises myocardial function in patients, who postoperatively develop haemodynamically significant arrhythmias, and helps maintain a stable cardiac rhythm.
However, placement of epicardial pacing leads is not without risk. In the short term, function deteriorates mainly due to the inflammation around the surfaces of the myocardium and the leads. Daily checking of the pacemaker’s pacing (the depolarisation of the atria or ventricles resulting from an impulse delivered from the generator down a lead to the heart) and sensing (the detection by the generator of intrinsic atrial or ventricular depolarisation signals) parameters after cardiac surgery is therefore paramount.
Pacemaker under-sensing occurs when the pacemaker fails to detect spontaneous myocardial depolarisation-as probably happened in our patient-and is a common reason for the appearance of inappropriate ventricular pacing spikes. When the spikes fall on the T wave, then the patient may develop a tachyarrhythmia-including polymorphic ventricular tachycardia. Increasing the pacemaker rate above that of the intrinsic heart rate may reduce the possibility of production of inappropriate ventricular pacing spikes.
(3). Sadoff J. Final Analysis of Efficacy and Safety of Single-Dose Ad26.COV2.S. 2022.
The Ad26.COV2.S vaccine (Johnson & Johnson-Janssen) is a recombinant, replication-incompetent human adenovirus type 26 (Ad26) vector encoding a full-length, membrane-bound severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein in a prefusion stabilized conformation.1,2 Primary analysis of the phase 3 ENSEMBLE trial, performed when preset criteria had been met and conducted during the early emergence of variants and for a median follow-up of 58 days, showed 66.9% efficacy against moderate to severe-critical (i.e., severe or critical) coronavirus disease 2019 (Covid-19) and greater than 85% efficacy against severe-critical disease.3 Here, we report the final analysis of the double-blind phase of ENSEMBLE, which was conducted in accordance with the protocol when data for more than 90% of the participants had been unblinded.
Background
The Ad26.COV2.S vaccine was highly effective against severe-critical coronavirus disease 2019 (Covid-19), hospitalization, and death in the primary phase 3 efficacy analysis.
Methods
We conducted the final analysis in the double-blind phase of our multinational, randomized, placebo-controlled trial, in which adults were assigned in a 1:1 ratio to receive single-dose Ad26.COV2.S (5×1010 viral particles) or placebo. The primary end points were vaccine efficacy against moderate to severe-critical Covid-19 with onset at least 14 days after administration and at least 28 days after administration in the per-protocol population. Safety and key secondary and exploratory end points were also assessed.
Results
Median follow-up in this analysis was 4 months; 8940 participants had at least 6 months of follow-up. In the per-protocol population (39,185 participants), vaccine efficacy against moderate to severe-critical Covid-19 at least 14 days after administration was 56.3% (95% confidence interval [CI], 51.3 to 60.8; 484 cases in the vaccine group vs. 1067 in the placebo group); at least 28 days after administration, vaccine efficacy was 52.9% (95% CI, 47.1 to 58.1; 433 cases in the vaccine group vs. 883 in the placebo group). Efficacy in the United States, primarily against the reference strain (B.1.D614G) and the B.1.1.7 (alpha) variant, was 69.7% (95% CI, 60.7 to 76.9); efficacy was reduced elsewhere against the P.1 (gamma), C.37 (lambda), and B.1.621 (mu) variants. Efficacy was 74.6% (95% CI, 64.7 to 82.1) against severe-critical Covid-19 (with only 4 severe-critical cases caused by the B.1.617.2 [delta] variant), 75.6% (95% CI, 54.3 to 88.0) against Covid-19 leading to medical intervention (including hospitalization), and 82.8% (95% CI, 40.5 to 96.8) against Covid-19-related death, with protection lasting 6 months or longer. Efficacy against any severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection was 41.7% (95% CI, 36.3 to 46.7). Ad26.COV2.S was associated with mainly mild-to-moderate adverse events, and no new safety concerns were identified.
Conclusions
A single dose of Ad26.COV2.S provided 52.9% protection against moderate to severe-critical Covid-19. Protection varied according to variant; higher protection was observed against severe Covid-19, medical intervention, and death than against other end points and lasted for 6 months or longer.
(4). Altarawneh HN. Protection against the Omicron Variant from Previous SARS-CoV-2 Infection. 2022.
To the Editor
Natural infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) elicits strong protection against reinfection with the B.1.1.7 (alpha), B.1.351 (beta),1 and B.1.617.2 (delta) variants. However, the B.1.1.529 (omicron) variant harbors multiple mutations that can mediate immune evasion. We estimated the effectiveness of previous infection in preventing symptomatic new cases caused by omicron and other SARS-CoV-2 variants in Qatar. In this study, we extracted data regarding coronavirus disease 2019 (Covid-19) laboratory testing, vaccination, clinical infection data, and related demographic details from the national SARS-CoV-2 databases, which include all results of polymerase-chain-reaction (PCR) testing, vaccinations, and hospitalizations and deaths for Covid-19 in Qatar since the start of the pandemic.
The effectiveness of previous SARS-CoV-2 infection in preventing reinfection was defined as the proportional reduction in susceptibility to infection among persons who had recovered from infection as compared with those who had not been infected. Previous SARS-CoV-2 infection was defined as a positive result on PCR assay at least 90 days before a new positive PCR finding. We used a test-negative, case-control study design to assess the effectiveness of previous infection in preventing reinfection on the basis of a method that had recently been investigated and validated for derivation of robust estimates for such comparisons In addition, we performed sensitivity analyses that included adjustment for vaccination status and that excluded vaccinated persons from the analysis. Case patients (defined as persons with positive PCR results) and controls (defined as persons with negative PCR results) were matched according to sex, 10-year age group, nationality, and calendar time of PCR testing to control for known differences in the risk of exposure to SARS-CoV-2 infection in Qatar.
To ensure that epidemiologically relevant reinfections were considered in the analysis, only documented infections with a PCR cycle threshold (Ct) value of 30 or less were included as cases in our study. (Reinfection often occurs with negligible symptoms and high Ct values, indicating reduced epidemiologic significance. We also estimated the effectiveness of previous infection in preventing hospitalization or death caused by reinfection.
The selection of the study population for various analyses is shown in Figures S1 through S4 and the population characteristics in Tables S1 and S2. The overall study population was broadly representative of the total population of Qatar (Table S3), with a median age of 31 to 35 years across the study samples. The median interval between previous infection and PCR testing among cases and controls was 279 days (interquartile range [IQR], 194 to 313) for analysis of the alpha variant, 285 days (IQR, 213 to 314) for analysis of the beta variant, 254 days (IQR, 159 to 376) for analysis of the delta variant, and 314 days (IQR, 268 to 487) for analysis of the omicron variant.
Effectiveness of Previous Infection with SARS-CoV-2 against Symptomatic Reinfection, According to Variant:
The effectiveness of previous infection in preventing reinfection was estimated to be 90.2% (95% confidence interval [CI], 60.2 to 97.6) against the alpha variant, 85.7% (95% CI, 75.8 to 91.7) against the beta variant, 92.0% (95% CI, 87.9 to 94.7) against the delta variant, and 56.0% (95% CI, 50.6 to 60.9) against the omicron variant. Sensitivity analyses confirmed the study results, as expected for this study design, which is robust regardless of the approach that is used to control for vaccine-induced immunity. An additional analysis that was adjusted for the interval since previous infection also confirmed the study results.
Among the patients with reinfection, progression to severe Covid-19 occurred in one patient with the alpha variant, in two patients with the beta variant, in no patients with the delta variant, and in two patients with the omicron variant. None of the reinfections progressed to critical or fatal Covid-19. The effectiveness with respect to severe, critical, or fatal Covid-19 was estimated to be 69.4% (95% CI, -143.6 to 96.2) against the alpha variant, 88.0% (95% CI, 50.7 to 97.1) against the beta variant, 100% (95% CI, 43.3 to 100) against the delta variant, and 87.8% (95% CI, 47.5 to 97.1) against the omicron variant. (For the delta variant, the calculation of the 95% confidence interval is clarified in a footnote in Table 1.) Limitations of the estimations (e.g., the relatively young population of Qatar) are discussed in Section S1.
Overall, in a national database study in Qatar, we found that the effectiveness of previous infection in preventing reinfection with the alpha, beta, and delta variants of SARS-CoV-2 was robust (at approximately 90%), findings that confirmed earlier estimates. Such protection against reinfection with the omicron variant was lower (approximately 60%) but still considerable. In addition, the protection of previous infection against hospitalization or death caused by reinfection appeared to be robust, regardless of variant.
(5). Bernal AJ. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N Engl J Med. 2022;386:509-20.
Background
New treatments are needed to reduce the risk of progression of coronavirus disease 2019 (Covid-19). Molnupiravir is an oral, small-molecule antiviral prodrug that is active against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Methods
We conducted a phase 3, double-blind, randomized, placebo-controlled trial to evaluate the efficacy and safety of treatment with molnupiravir started within 5 days after the onset of signs or symptoms in nonhospitalized, unvaccinated adults with mild-to-moderate, laboratory-confirmed Covid-19 and at least one risk factor for severe Covid-19 illness. Participants in the trial were randomly assigned to receive 800 mg of molnupiravir or placebo twice daily for 5 days. The primary efficacy end point was the incidence hospitalization or death at day 29; the incidence of adverse events was the primary safety end point. A planned interim analysis was performed when 50% of 1550 participants (target enrollment) had been followed through day 29.
Results
A total of 1433 participants underwent randomization; 716 were assigned to receive molnupiravir and 717 to receive placebo. With the exception of an imbalance in sex, baseline characteristics were similar in the two groups. The superiority of molnupiravir was demonstrated at the interim analysis; the risk of hospitalization for any cause or death through day 29 was lower with molnupiravir (28 of 385 participants [7.3%]) than with placebo (53 of 377 [14.1%]) (difference, -6.8 percentage points; 95% confidence interval [CI], -11.3 to -2.4; P=0.001). In the analysis of all participants who had undergone randomization, the percentage of participants who were hospitalized or died through day 29 was lower in the molnupiravir group than in the placebo group (6.8% [48 of 709] vs. 9.7% [68 of 699]; difference, -3.0 percentage points; 95% CI, -5.9 to -0.1). Results of subgroup analyses were largely consistent with these overall results; in some subgroups, such as patients with evidence of previous SARS-CoV-2 infection, those with low baseline viral load, and those with diabetes, the point estimate for the difference favored placebo. One death was reported in the molnupiravir group and 9 were reported in the placebo group through day 29. Adverse events were reported in 216 of 710 participants (30.4%) in the molnupiravir group and 231 of 701 (33.0%) in the placebo group.
Conclusions
Early treatment with molnupiravir reduced the risk of hospitalization or death in at-risk, unvaccinated adults with Covid-19.
(6). Schmidt F. Plasma Neutralization of the SARS-CoV-2 Omicron Variant. N Engl J Med 2022;386:599-601.
The newly emerged B.1.1.159 (omicron) variant of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has a large number of changes – 32 – in its spike protein relative to that of the original virus (Wuhan-hu-1), particularly in the receptor-binding domain and the N-terminal domain, the primary targets of neutralizing antibodies. Previously, we showed that approximately 20 changes introduced into a synthetic polymutant spike protein (PMS20) are sufficient for substantial evasion of the polyclonal neutralizing antibodies elicited in the majority of persons who have recovered from coronavirus disease 2019 (Covid-19) or have received two doses of an mRNA vaccine. Of note, several changes in the PMS20 spike protein are the same as or similar to changes in the omicron variant.
We measured neutralizing antibody titers against Wuhan-hu-1, PMS20, and omicron spike pseudotypes in 169 plasma specimens from 47 persons with diverse exposures to SARS-CoV-2 antigens through infection, vaccination, or. In plasma specimens obtained at approximately 1 month and 6 months after infection from persons who had recovered from Covid-19, the 50% neutralization titer (NT50) values were a mean (±SD) of 60±47 and 37±27 times lower for PMS20 than for Wuhan-hu-1, respectively, and 58±51 and 32±23 times lower for omicron than for Wuhan-hu-1. Similarly, plasma specimens obtained from different persons in the same cohort 1 year after infection had NT50 values that were 34±24 times lower for PMS20 and 43±23 times lower for omicron than for Wuhan-hu-1.
In plasma specimens from persons who had received two doses of an mRNA vaccine (BNT162b2 [Pfizer-BioNTech] or mRNA-1273 [Moderna]) 1.3 months before sampling, the NT50 values were 187±24 times lower for PMS20 and 127±66 times lower for omicron than for Wuhan-hu-1. At 5 months after vaccination, the neutralization potency was 58±23 times lower for PMS20 and 27±17 times lower for omicron. Many plasma specimens from recipients of the single-dose Ad26.COV2.S vaccine (Johnson & Johnson-Janssen), obtained 1 or 5 months after vaccination, lacked detectable neutralizing activity against PMS20 or omicron, which precluded a meaningful quantitative assessment of variant-specific differences.
Wuhan-hu-1, PMS20, and Omicron Plasma Neutralizing Titers:
Of note, however, vaccination of persons who had recovered from Covid-19 or administration of a third dose of an mRNA vaccine to vaccinated persons at least 6 months after the second dose of an mRNA vaccine led to a substantial gain in neutralizing activity against PMS20 and omicron. Specifically, after vaccination in persons who had previously been infected with SARS-CoV-2, the NT50 values were 238 times, 214 times, and 154 times greater for Wuhan-hu-1, PMS20, and omicron pseudotypes, respectively, than the prevaccination convalescent-phase titers in the same persons. For those who had received two doses of an mRNA vaccine approximately 6 months earlier and then received a third dose of an mRNA vaccine approximately 1 month before sampling, the NT50 values after the booster dose were 26 times greater for Wuhan-hu-1, 35 times greater for PMS20, and 38 times greater for omicron. Neutralizing titers against omicron were substantial (ranging from 1411 to 56,537) in all persons who had had Covid-19 and were then vaccinated and in those who had received three doses of an mRNA vaccine, but titers were low or undetectable in many unvaccinated persons who had had Covid-19 and in recipients of only two doses of an mRNA vaccine.
Although these findings indicate that the omicron variant shows an unprecedented degree of neutralizing antibody escape, they also suggest that boosting and promoting affinity maturation of antibodies in persons who have previously been infected or vaccinated,with the use of existing Wuhan-hu-1-based vaccine immunogens, will provide additional protection against infection with the omicron variant and subsequent disease.
(7). Hammond J. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. 2022.
Background
Nirmatrelvir is an orally administered severe acute respiratory syndrome coronavirus 2 main protease (Mpro) inhibitor with potent pan-human-coronavirus activity in vitro.
Methods
We conducted a phase 2-3 double-blind, randomized, controlled trial in which symptomatic, unvaccinated, nonhospitalized adults at high risk for progression to severe coronavirus disease 2019 (Covid-19) were assigned in a 1:1 ratio to receive either 300 mg of nirmatrelvir plus 100 mg of ritonavir (a pharmacokinetic enhancer) or placebo every 12 hours for 5 days. Covid-19-related hospitalization or death from any cause through day 28, viral load, and safety were evaluated.
Results
A total of 2246 patients underwent randomization; 1120 patients received nirmatrelvir plus ritonavir (nirmatrelvir group) and 1126 received placebo (placebo group). In the planned interim analysis of patients treated within 3 days after symptom onset (modified intention-to treat population, comprising 774 of the 1361 patients in the full analysis population), the incidence of Covid-19-related hospitalization or death by day 28 was lower in the nirmatrelvir group than in the placebo group by 6.32 percentage points (95% confidence interval [CI], -9.04 to -3.59; P<0.001; relative risk reduction, 89.1%); the incidence was 0.77% (3 of 389 patients) in the nirmatrelvir group, with 0 deaths, as compared with 7.01% (27 of 385 patients) in the placebo group, with 7 deaths. Efficacy was maintained in the final analysis involving the 1379 patients in the modified intention-to-treat population, with a difference of -5.81 percentage points (95% CI, -7.78 to -3.84; P<0.001; relative risk reduction, 88.9%). All 13 deaths occurred in the placebo group. The viral load was lower with nirmaltrelvir plus ritonavir than with placebo at day 5 of treatment, with an adjusted mean difference of -0.868 log10 copies per milliliter when treatment was initiated within 3 days after the onset of symptoms. The incidence of adverse events that emerged during the treatment period was similar in the two groups (any adverse event, 22.6% with nirmatrelvir plus ritonavir vs. 23.9% with placebo; serious adverse events, 1.6% vs. 6.6%; and adverse events leading to discontinuation of the drugs or placebo, 2.1% vs. 4.2%). Dysgeusia (5.6% vs. 0.3%) and diarrhea (3.1% vs. 1.6%) occurred more frequently with nirmatrelvir plus ritonavir than with placebo.
Conclusions
Treatment of symptomatic Covid-19 with nirmatrelvir plus ritonavir resulted in a risk of progression to severe Covid-19 that was 89% lower than the risk with placebo, without evident safety concerns.
(8). Anderson LJ. The Challenge of Respiratory Syncytial Virus Human Challenge Studies. N Engl J Med. 2022;386:696-7.
The development of drugs for the treatment of respiratory syncytial virus (RSV), a major cause of respiratory disease in young children and high-risk adults, is a high priority. In this study Ahmad et al. report the results of a human challenge trial of EDP-938, a small molecule targeting the RSV nucleoprotein (N protein).
Challenge studies with predictable virus shedding and mild-to-moderate symptoms affecting the upper respiratory tract in study participants have been used to evaluate the efficacy of antiviral drugs and to collect data that would support the conduct of subsequent clinical trials.
Multiple drugs targeting the RSV fusion (F) protein, the RNA-dependent RNA polymerase large (L) protein, and the N protein have been evaluated in challenge studies, but the results were disappointing in follow-up clinical trials. For example, the use of presatovir (GS-5806), a small-molecule fusion inhibitor targeting the F protein, resulted in impressive reductions in the viral load and the severity of symptoms in a human challenge study but was ineffective in clinical trials. The RSV attachment (G) protein has also been used as a target but only in preclinical treatment studies. Blocking G protein activity might have both an antiviral and antiinflammatory effect.
In the two-part trial 115 healthy young adult volunteers in part 1 and 63 in part 2 were intranasally inoculated with the Memphis 37b strain of RSV and then began an oral regimen of EDP-938 or placebo.
Various combinations of loading doses (400 to 600 mg) followed by maintenance doses (once-daily doses of 600 mg or 300 mg or twice-daily doses of 300 mg or 200 mg) of EDP-938 or placebo were given for 5 days starting on the day of the first positive polymerase-chain-reaction (PCR) test or starting on day 5 after inoculation if the virus had not yet been detected.
The primary end point was the area under the curve (AUC) of the viral load, as measured by PCR assay; the key secondary end point was the AUC of the total symptom score (symptoms were assessed by the participants); and other secondary end points were the AUC of the viral load, as measured by cell-based infectivity assay, and nasal mucus weight. End-point data were recorded each day for 10 days, beginning with the day of the first dose. A total of 86 volunteers in part 1 and 38 in part 2 met the criteria for RSV infection and were included in the end-point efficacy analyses.
In both parts of the trial, the authors found that the AUC of the viral load, as measured by PCR assay and expressed as hours×log10 copies per milliliter, was lower in the EDP-938 groups than in the placebo group (P<0.001) (e.g., in part 1, the AUC of the viral load was 204.0 in the 600-mg once-daily group, 217.7 in the 300-mg twice-daily group, and 790.2 in the placebo group, corresponding with a viral-load AUC in the 600-mg once-daily group that was 3.9 times as low as that in the placebo group and a viral-load AUC in the 300-mg twice-daily group that was 3.6 times as low as that in the placebo group). The AUC of the mean total symptom score and the mucus weight were also lower in all the EDP-938 groups than in the placebo group.
No apparent safety signals were identified with EDP-938, and the drug had a relatively long half-life of approximately 14 hours. These results support moving forward with clinical trials.
What have we learned from this challenge study?
First, the trial design was patterned after earlier randomized RSV-challenge drug trials that used established outcome measures and had encouraging results that supported moving forward to clinical trials. However, positive results of treatment in previous challenge studies have not been followed by success in later clinical trials. It is likely that the difference in the timing of treatment contributes to the discrepancy between challenge studies and clinical trials.
In challenge studies, the drug is given early, whereas in clinical trials of natural RSV infection, the drug is given when viral titers have peaked, since patients generally present for care 3 to 8 days after symptom onset (i.e., 5 to 10 days after infection, with an assumption of 2 days between the earliest onset of PCR positivity and the onset of symptoms). The rapid, anamnestic immune response in adults who had been primed with RSV might outcompete the antiviral effects of a drug in clinical trials but is less likely to do so in challenge studies. Even in infants who had not been previously infected with RSV, viral titers are in decline at the time of hospitalization, probably because of effective viral clearance by rapid innate immune responses.
Thus, disease caused by active virus replication would more likely be affected by treatment in challenge studies than in clinical trials.
Another potential factor is a host immune response that contributes to disease through inflammatory mediators. It is possible that later in the course of infection, these responses become less dependent on virus replication, and an antiviral effect is less likely to affect disease in clinical trials. Patients who may be more likely to have a response to later treatment are those with immunosuppression, who often have prolonged virus shedding and progressive disease.
The need for safe and effective treatments for RSV remains high and unfulfilled. It is hoped that EDP-938 or other products will prove to be successful and meet this need.
It is, however, possible that new approaches are needed. For example, will treatment require both antiviral and antiinflammatory activity to be effective, and would starting treatment after symptom onset add value to challenge studies?
A challenge in challenge studies is how best to design and use them to plan clinical trials that would most efficiently evaluate new treatments for RSV disease.
(9). Frías JP. Tirzepatide versus Semaglutide Once Weekly in Type 2 Diabetes. N Engl J Med 2022; 386:e17.
In the SURPASS-2 trial (A Study of Tirzepatide [LY3298176] versus Semaglutide Once Weekly as Add-on Therapy to Metformin in Participants with Type 2 Diabetes), Frías et al. (Aug. 5 issue)1 report that treatment with tirzepatide, a dual glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 (GLP-1) receptor agonist, resulted in better glycemic control and greater weight loss than semaglutide in patients with type 2 diabetes mellitus.
(10). Luc Montagnier, Nobel-Winning Virologist Who Discovered HIV, Dies at 89.
The French virologist Luc Montagnier, PhD, who shared the 2008 Nobel Prize in Medicine for his work in isolating the human immunodeficiency virus, has died at age 89.
Dr Luc Montagnier died on Tuesday in the Paris suburb of Neuilly-sur-Seine.
Montagnier and colleague Francoise Barre-Sinoussi, PhD, shared the Nobel for their work at the Pasteur Institute in Paris. Their achievement paved the way for HIV tests and antiretroviral drugs that allow patients to manage the virus as a chronic illness.
While Montagnier’s vital early discoveries on AIDS were celebrated, he was later dismissed by scientists “for his increasingly outlandish theories,” notably, statements related to COVID-19.
Among his statements were that the SARS-CoV-2 virus was laboratory-made and that vaccines were responsible for the appearance of variants.
He also suggested that autism is caused by infection and set up much-criticized experiments to prove it. He claimed antibiotics could cure the condition.
“And he believed that anyone with a good immune system could fight off HIV with the right diet,”
“He was always controversial, but I had the greatest respect for the team he assembled,” Donald P. Francis, who directed the AIDS laboratory at the Centers for Disease Control and Prevention in the early days of the AIDS epidemic.”
Montagnier had a bitter rivalry with US scientist Robert Gallo in identifying HIV. Montagnier sued Gallo for using his discovery for a US patent.
Former President Ronald Reagan and former French Prime Minister Jacques Chirac entered the fray, with both sides claiming a share of the credit.
The suit was settled out of court, mediated by Jonas Salk, who had years earlier been involved in a similar battle with Albert Sabin over the polio vaccine.
Montagnier and Gallo are co-credited with discovering that HIV causes AIDS. Although the Nobel jury made no mention of Gallo in its citation, in 1986, Montagnier and Gallo shared the Lasker Award – the US equivalent of the Nobel – with Myron Essex, PhD, of the Harvard School of Public Health. Montagnier was credited with discovering the virus and Gallo for linking it to AIDS.
How the HIV Discovery Began:
The path to discover HIV started in Paris in the Viral Oncology Unit at the Pasteur Institute on January 3, 1983, according to The New York Times. On that day, Montagnier received a piece of lymph node that had been removed from a 33-year-old man with AIDS.
At that point there was no known cause, no known tests, and no known treatments for AIDS.
Montagnier was an expert in retroviruses, and many physicians were beginning to suspect AIDS was caused by a retrovirus – an RNA virus that replicates by inserting a DNA copy of its genome into a host cell.
Montagnier’s team found in the lymph-node sample a retrovirus never seen before. They first named it LAV, for lymphadenopathy-associated virus. The team reported the landmark results in the May 20, 1983, issue of the journal Science, concluding that further studies were needed to prove that LAV caused AIDS.
The next year, the laboratory run by Gallo, at the National Institutes of Health, published four articles in one issue of Science confirming the link between a retrovirus and AIDS.
Tributes include one from American physicist Richard M. Fleming, saying, “A great light has gone out in the world today.”
