Journal scan: A review of 10 recent papers of immediate clinical significance, harvested from major international journals
From the desk of the Editor-in-Chief
(1). Mahase E. COVID-19: UK approves monoclonal antibody sotrovimab for over 12s at high risk. BMJ 2021;375:n2990.
The UK’s medicines regulator has approved a second monoclonal antibody, sotrovimab, for the treatment of people over 12 years with mild to moderate COVID-19 who are at high risk of developing severe disease.
The Medicines and Healthcare Products Regulatory Agency’s decision was based on clinical trial data showing that a single dose of sotrovimab, which is given as an intravenous infusion over 30 minutes, reduced the risk of hospital admission and death by 79% in high risk adults with symptomatic COVID-19.
The MHRA concluded from the trial findings that sotrovimab is most effective when taken during the early stages of infection and so is recommended for use within five days of symptoms starting.
Developed by GSK and Vir Biotechnology, sotrovimab is a single monoclonal antibody that works by binding to the SARS-CoV-2 spike protein, thereby preventing the virus from attaching to and entering human cells.
Sotrovimab has been authorised for use in people with at least one risk factor for developing severe illness, such as obesity, being aged over 60 years, diabetes mellitus, and heart disease.
It is being claimed by the manufacturer that it is effective against Omicron
(2). Dyer O. COVID-19: FDA expert panel recommends authorising molnupiravir but also voices concerns. BMJ 2021;375:n2984.
US Food and Drug Administration has granted an emergency authorisation to Merck’s molnupiravir (Lagevrio), an antiviral for the outpatient treatment of COVID-19.
The vote on molnupiravir was unusual for its narrow margin and the many reservations expressed by panellists who voted to recommend the drug. The nine-hour meeting was broadcast live.
Molnupiravir was claimed by Merck as having reduced hospital admissions by 50% in a phase III trial. That was the benefit cited by regulators in the UK, the first country to authorise the drug’s use last month. But the data presented to the FDA panel showed only a 30% reduction in admissions, a change that several panellists said was poorly explained.
The US has already authorised three monoclonal antibody cocktails, which have shown efficacy above 60% in preventing admission, and the FDA does not generally approve drugs that are less efficacious than those already in use.
Regeneron, maker of the most used monoclonal, REGEN-COV, said this week that early testing suggested that its efficacy was reduced against the omicron variant.
Molnupiravir is a course of pills that can be taken at home and costs one third of the price of REGEN-COV.
Several panellists who voted to recommend molnupiravir added qualifications and caveats or described it as a stopgap measure.
An FDA panel is considering another new COVID antiviral, Pfizer’s Paxlovid. Pfizer has claimed that it is 89% effective in reducing hospital admission.
New data darken outlook
Genetic danger
Molnupiravir attacks the coronavirus by triggering an accumulation of errors in the viral genome, and there have been concerns that it might alter human genes. But the panellists were shown data from animal studies that suggested low risk to adult humans at therapeutic doses. The drug will not be authorised in anyone who is pregnant or lactating, and its paediatric future is not clear.
Panellists were far less reassured about the drug’s impact on coronavirus genes. While its mutagenic effect is designed to stop the virus replicating, mutations that make it more infective or vaccine resistant are also possible. In molnupiravir’s phase II trial, 72 structural nucleotide changes to the spike protein were found in the treatment arm, compared with nine in the placebo arm.
Several of these changes were similar to those seen in major variants including delta, FDA researchers told the panel. But they stopped short of concluding that molnupiravir would increase the risk of new variants, noting that the coronavirus spike protein was already mutating in nature.
James Hildreth, president of Meharry Medical College in Tennessee, who ultimately voted no, said that Merck should do more to quantify the risk. “Even if the probability is very low, 1 in 10 000 or 100 000, that this drug would induce an escape mutant which the vaccines we have do not cover, that would be catastrophic for the whole world,” he said.
(3). Edouard LF, et al. Timing of dialysis initiation to reduce mortality and cardiovascular events in advanced chronic kidney disease: nationwide cohort study. BMJ 2021;375:e066306.
Abstract
Objective: To identify the optimal estimated glomerular filtration rate (eGFR) at which to initiate dialysis in people with advanced chronic kidney disease.
Design: Nationwide observational cohort study.
Setting: National Swedish Renal Registry of patients referred to nephrologists.
Participants: Patients had a baseline eGFR between 10 and 20 ml/min/1.73 m2 and were included between 1 January 2007 and 31 December 2016, with follow-up until 1 June 2017.
Main outcome measures: The strict design criteria of a clinical trial were mimicked by using the cloning, censoring, and weighting method to eliminate immortal time bias, lead time bias, and survivor bias. A dynamic marginal structural model was used to estimate adjusted hazard ratios and absolute risks for five year all-cause mortality and major adverse cardiovascular events (composite of cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke) for 15 dialysis initiation strategies with eGFR values between 4 and 19 ml/min/1.73 m2 in increments of 1 ml/min/1.73 m2. An eGFR between 6 and 7 ml/min/1.73 m2 (eGFR6-7) was taken as the reference.
Results: Among 10,290 incident patients with advanced chronic kidney disease (median age 73 years; 3739 (36%) women; median eGFR 16.8 ml/min/1.73 m2), 3822 started dialysis, 4160 died, and 2446 had a major adverse cardiovascular event. A parabolic relation was observed for mortality, with the lowest risk for eGFR15-16. Compared with dialysis initiation at eGFR6-7, initiation at eGFR15-16 was associated with a 5.1% (95% confidence interval 2.5 to 6.9%) lower absolute five-year mortality risk and 2.9% (0.2 to 5.5%) lower risk of a major adverse cardiovascular event, corresponding to hazard ratios of 0.89 (95% confidence interval 0.87 to 0.92) and 0.94 (0.91 to 0.98), respectively. This 5.1% absolute risk difference corresponded to a mean postponement of death of 1.6 months over five years of follow-up. However, dialysis would need to be started four years earlier. When emulating the intended strategies of the Initiating Dialysis Early and Late (IDEAL) trial (eGFR10-14v eGFR5-7) and the achieved eGFRs in IDEAL (eGFR7-10v eGFR5-7), hazard ratios for all-cause mortality were 0.96 (0.94 to 0.99) and 0.97 (0.94 to 1.00), respectively, which are congruent with the findings of the randomised IDEAL trial.
Conclusions: Very early initiation of dialysis was associated with a modest reduction in mortality and cardiovascular events. For most patients, such a reduction may not outweigh the burden of a substantially longer period spent on dialysis.
(4). Al-Matar SH. Ultrashort case report in the Lancet! Fever, abdominal pain, serositis, arthralgia, hearing loss, proteinuria, and a family history: Muckle Wells syndrome. Lancet 2021;398(10316):P2101.
A 53-year-old man presented with a 3-week history of fevers, intermittent abdominal pain, anorexia, arthralgias, fatigue, generalised body swelling, and 6.8 kg of unintentional weight loss.
The patient had a history of bilateral sensorineural hearing loss diagnosed when he was around 12 years old.
On examination, we found the patient to be cachexic and wheelchair bound from anasarca; he had bilateral conjunctivitis and a non-tender abdomen with ascites.
Initial laboratory investigations showed a neutrophilic leukocytosis: his white blood cell count was 36.20 × 109 per L and his neutrophil count was 33.86 × 109 per L. He had a normocytic anaemia: his haemoglobin concentration was 94 g/L and his mean corpuscular volume was 89.5 fL. The serum C-reactive protein concentration was elevated at 141.31 mg/L (normal 0-5); the serum IgA concentration was also raised at 9.66 g/L (normal 0.8-4.0). The patient’s serum albumin concentration was low at 18 g/L (normal 38-52); renal and liver function tests showed no abnormalities. Subsequent investigations identified nephrotic range proteinuria, with normal titres of antinuclear antibodies, antineutrophil cytoplasmic antibodies, and C3 and C4, and normal serum calcium concentration.
Initial imaging showed a moderately sized left-sided pleural effusion, abdominal ascites, and retroperitoneal, iliac, and inguinal lymphadenopathy.
Investigations for possible infections, cardiac diseases, and malignancies were all negative (appendix). On further history taking, the patient reported similar recurring episodes since his childhood without any clear potential precipitants—although occasionally, he had a cold-induced urticarial rash. Notably, the patient’s maternal grandmother, a maternal aunt, and his mother—who had haemodialysis for renal amyloidosis—had similar episodes.
Given the recurrent episodes of fever, abdominal pain, serositis, and arthralgia, with hearing loss, proteinuria, and strong family history suggesting the vertical transmission of an autosomal dominant trait, we suspected the rare, genetic, autoinflammatory disorder, Muckle Wells syndrome (MWS). Genetic sequencing showed a heterozygous mutation (c.1049C>T) in the NLRP3 gene—a mutation on the long arm of chromosome 1 previously reported in multiple individuals with MWS.
We commenced treatment with canakinumab, a monoclonal antibody targeting interleukin 1β (IL-1β), at a dose of 150 mg subcutaneously every 8 weeks, and the patient’s inflammatory manifestations of the disorder swiftly resolved.
In MWS, gain-of-function mutations in NLRP3 dysregulate the inflammasome complex, enhancing IL-1β activity. Despite its genetic basis, the disorder can be seen in adults. Recurring inflammation in the inner ear causes the sensorineural deafness; accumulation of amyloid fibrils in various organs—including the kidneys—results in amyloid A amyloidosis. Early recognition and treatment are key in preventing these sequelae.
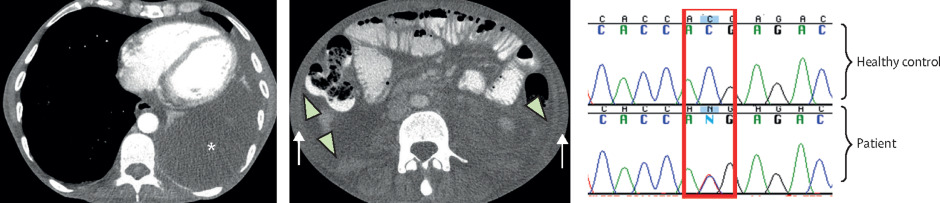
(A) CT scan of the thorax shows a large left-sided pleural effusion. (B) CT scan of the abdomen shows ascites (arrowhead) and abdominal wall soft tissue anasarca (arrow). (C) Results of sequencing show a heterozygous C/T mutation in the patient compared to a healthy control (red box). *Indicates the effusion.
(5). Khanna S. Ultra-short case report in the NEJM. Ectopic Tooth in the Nose. 2021.
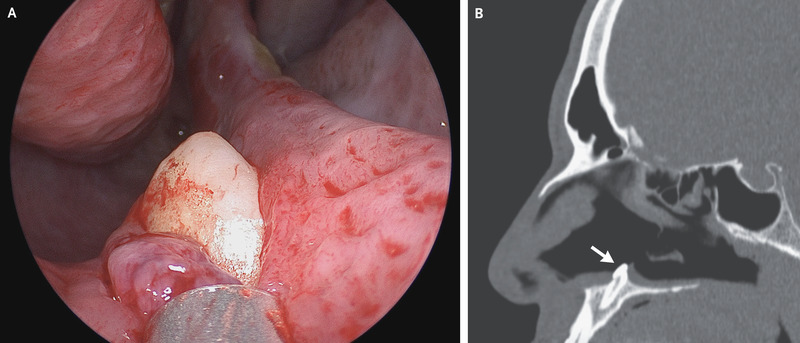
A 38-year-old man presented to the otolaryngology clinic with difficulty in breathing through his right nostril, which he said had been ongoing for several years. He reported no history of facial trauma or craniofacial abnormalities. Physical examination of the nose showed a septal deviation, calcified septal spurs, and a 2-cm perforation in the posterior septum. On rhinoscopy, a hard, nontender, white mass was observed in the floor of the right nostril (Panel A). Computed tomography of the paranasal sinuses showed a well-defined, radiodense mass consistent with an inverted ectopic tooth in the nasal cavity (Panel B, arrow), which was thought to explain the obstructive symptoms and septal perforation. The tooth was removed during oral and otolaryngologic surgery by means of an intranasal approach and measured 14 mm in length. There were no postoperative complications. At follow-up 3 months after surgery, the patient’s symptoms of nasal obstruction had resolved.
(6). Mevorach D. Myocarditis after BNT162b2 mRNA Vaccine against COVID-19 in Israel. N Engl J Med. 2021;385:2140-9.
Abstract
Background
Approximately 5.1 million Israelis had been fully immunized against coronavirus disease 2019 (COVID-19) after receiving two doses of the BNT162b2 messenger RNA vaccine (Pfizer-BioNTech) by May 31, 2021. After early reports of myocarditis during adverse events monitoring, the Israeli Ministry of Health initiated active surveillance.
Methods
We retrospectively reviewed data obtained from December 20, 2020, to May 31, 2021, regarding all cases of myocarditis and categorized the information using the Brighton Collaboration definition. We analyzed the occurrence of myocarditis by computing the risk difference for the comparison of the incidence after the first and second vaccine doses (21 days apart); by calculating the standardized incidence ratio of the observed-to-expected incidence within 21 days after the first dose and 30 days after the second dose, independent of certainty of diagnosis; and by calculating the rate ratio 30 days after the second dose as compared with unvaccinated persons.
Among 304 persons with symptoms of myocarditis, 21 had received an alternative diagnosis. Of the remaining 283 cases, 142 occurred after receipt of the BNT162b2 vaccine; of these cases, 136 diagnoses were definitive or probable. The clinical presentation was judged to be mild in 129 recipients (95%); one fulminant case was fatal. The overall risk difference between the first and second doses was 1.76 per 100,000 persons (95% confidence interval [CI], 1.33 to 2.19), with the largest difference among male recipients between the ages of 16 and 19 years (difference, 13.73 per 100,000 persons; 95% CI, 8.11 to 19.46). As compared with the expected incidence based on historical data, the standardized incidence ratio was 5.34 (95% CI, 4.48 to 6.40) and was highest after the second dose in male recipients between the ages of 16 and 19 years (13.60; 95% CI, 9.30 to 19.20). The rate ratio 30 days after the second vaccine dose in fully vaccinated recipients, as compared with unvaccinated persons, was 2.35 (95% CI, 1.10 to 5.02); the rate ratio was again highest in male recipients between the ages of 16 and 19 years (8.96; 95% CI, 4.50 to 17.83), with a ratio of 1 in 6637.
Conclusions
The incidence of myocarditis, although low, increased after the receipt of the BNT162b2 vaccine, particularly after the second dose among young male recipients. The clinical presentation of myocarditis after vaccination was usually mild.
(7). Weitz JI. Milvexian for the Prevention of Venous Thromboembolism. N Engl J Med. 2021;385:2161-72.
Abstract
Background
Factor XIa inhibitors for the prevention and treatment of venous and arterial thromboembolism may be more effective and result in less bleeding than conventional anticoagulants. Additional data are needed regarding the efficacy and safety of milvexian, an oral factor XIa inhibitor.
Methods
In this parallel-group, phase 2 trial, we randomly assigned 1242 patients undergoing knee arthroplasty to receive one of seven postoperative regimens of milvexian (25, 50, 100, or 200 mg twice daily or 25, 50, or 200 mg once daily) or enoxaparin (40 mg once daily). The primary efficacy outcome was venous thromboembolism (which was a composite of asymptomatic deep-vein thrombosis, confirmed symptomatic venous thromboembolism, or death from any cause). The principal safety outcome was bleeding.
Among the patients receiving milvexian twice daily, venous thromboembolism developed in 27 of 129 (21%) taking 25 mg, in 14 of 124 (11%) taking 50 mg, in 12 of 134 (9%) taking 100 mg, and in 10 of 131 (8%) taking 200 mg. Among those receiving milvexian once daily, venous thromboembolism developed in 7 of 28 (25%) taking 25 mg, in 30 of 127 (24%) taking 50 mg, and in 8 of 123 (7%) taking 200 mg, as compared with 54 of 252 patients (21%) taking enoxaparin. The dose-response relationship with twice-daily milvexian was significant (one-sided P < 0.001), and the 12% incidence of venous thromboembolism with twice-daily milvexian was significantly lower than the prespecified benchmark of 30% (one-sided P < 0.001). Bleeding of any severity occurred in 38 of 923 patients (4%) taking milvexian and in 12 of 296 patients (4%) taking enoxaparin; major or clinically relevant nonmajor bleeding occurred in 1 and 2%, respectively; and serious adverse events were reported in 2 and 4%, respectively.
Conclusions
Postoperative factor XIa inhibition with oral milvexian in patients undergoing knee arthroplasty was effective for the prevention of venous thromboembolism and was associated with a low risk of bleeding.
Oral anticoagulants are a mainstay for the prevention and treatment of venous and arterial thromboembolism. Although direct oral anticoagulants have replaced vitamin K antagonists for many indications, bleeding remains the major side effect. Fear of bleeding contributes to the underuse of anticoagulants in eligible patients with atrial fibrillation and to the inappropriate use of low-dose direct oral anticoagulant regimens. Therefore, the need for safer oral anticoagulants persists.
Factor XI is a promising target for the development of safer anticoagulants because it is an important driver of thrombus growth but plays a subsidiary part in hemostasis. Thus, patients with congenital factor XI deficiency are at lower risk for venous thromboembolism and ischemic stroke than those with normal factor XI levels but rarely have spontaneous bleeding.
The development of new anticoagulants usually starts with dose-finding studies involving patients undergoing elective knee arthroplasty, because efficacy can be objectively and efficiently assessed by using venography to determine the incidence of deep-vein thrombosis after surgery. In such patients, preoperative subcutaneous administration of an antisense oligonucleotide that reduces factor XI levels or postoperative factor XI inhibition with intravenous abelacimab, a factor XI-directed antibody, was superior to enoxaparin for the prevention of venous thromboembolism.
Milvexian is a selective factor XIa inhibitor that is rapidly absorbed after oral administration and has a half-life of approximately 12 h. In this proof-of-principle phase 2 trial (Antithrombotic Treatment with Factor XIa Inhibition to Optimize Management of Acute Thromboembolic Events in Total Knee Replacement [AXIOMATIC-TKR]), we compared the efficacy and safety of milvexian and enoxaparin in patients undergoing elective knee arthroplasty.
(8). Weinreich DM. REGEN-COV Antibody Combination and Outcomes in Outpatients with COVID-19. N Engl J Med. 2021;385:e81.
Abstract
Background
In the phase 1-2 portion of an adaptive trial, REGEN-COV, a combination of the monoclonal antibodies casirivimab and imdevimab, reduced the viral load and number of medical visits in patients with coronavirus disease 2019 (COVID-19). REGEN-COV has activity in vitro against current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern.
Methods
In the phase 3 portion of an adaptive trial, we randomly assigned outpatients with COVID-19 and risk factors for severe disease to receive various doses of intravenous REGEN-COV or placebo. Patients were followed through day 29. A prespecified hierarchical analysis was used to assess the end points of hospitalization or death and the time to resolution of symptoms. Safety was also evaluated.
Results
COVID-19-related hospitalization or death from any cause occurred in 18 of 1355 patients in the REGEN-COV 2400-mg group (1.3%) and in 62 of 1341 patients in the placebo group who underwent randomization concurrently (4.6%) (relative risk reduction [1 minus the relative risk], 71.3%; P<0.001); these outcomes occurred in 7 of 736 patients in the REGEN-COV 1200-mg group (1.0%) and in 24 of 748 patients in the placebo group who underwent randomization concurrently (3.2%) (relative risk reduction, 70.4%; P=0.002). The median time to resolution of symptoms was 4 days shorter with each REGEN-COV dose than with placebo (10 days vs. 14 days; P<0.001 for both comparisons). REGEN-COV was efficacious across various subgroups, including patients who were SARS-CoV-2 serum antibody-positive at baseline. Both REGEN-COV doses reduced viral load faster than placebo; the least-squares mean difference in viral load from baseline through day 7 was -0.71 log10 copies per milliliter (95% confidence interval [CI], -0.90 to -0.53) in the 1200-mg group and -0.86 log10 copies per milliliter (95% CI, -1.00 to -0.72) in the 2400-mg group. Serious adverse events occurred more frequently in the placebo group (4.0%) than in the 1200-mg group (1.1%) and the 2400-mg group (1.3%); infusion-related reactions of grade 2 or higher occurred in less than 0.3% of the patients in all groups.
Conclusions
REGEN-COV reduced the risk of COVID-19-related hospitalization or death from any cause, and it resolved symptoms and reduced the SARS-CoV-2 viral load more rapidly than placebo.
(9). Schievink WI. Spontaneous Intracranial Hypotension. N Engl J Med 2021;385:2173-8.
Spontaneous intracranial hypotension is a condition characterized by a lower-than-normal volume of cerebrospinal fluid (CSF) because of leakage of CSF through the dural membrane at one or multiple sites. The loss of CSF results in displacement of cerebral structures, causing headache and other neurologic symptoms. The term “spontaneous” in relation to the disorder is used to differentiate it from intracranial hypotension caused by CSF leaks of known cause, such as craniospinal trauma, spinal surgery, or most commonly, lumbar puncture or spinal anesthesia. Although CSF pressure in this disorder, as measured by manometry during lumbar puncture, may be normal, it is often lower than normal (with the normal value considered to be 6 to 25 cm of water, or 4.4 to 18.4 mm Hg), and the term “hypotension” continues to be used. Spontaneous intracranial hypotension assumes clinical importance as a treatable cause of headache and other manifestations, including dizziness, mental dullness, and behavioral changes, but the variability in clinical presentation makes diagnosis difficult.
A CSF leak within the spinal column is the most common identifiable cause of spontaneous intracranial hypotension. The mechanism underlying headache and neurologic manifestations is presumed to be downward displacement of cerebral structures and traction or distortion of pain-sensitive nerve endings in the cranial dura and its vasculature. Skull-base CSF leakage from the posterior cranial fossa into the soft tissues of the neck may rarely cause spontaneous intracranial hypotension, but skull-base CSF leaks resulting in CSF rhinorrhea or otorrhea have not been shown to cause spontaneous intracranial hypotension.
Summary
Spontaneous intracranial hypotension is a cause of headache and a variety of other neurologic manifestations. A causative CSF leak at the level of the spine can be detected in most patients, and several types of leaks have been identified. Epidural blood patching is successful in reducing or eliminating symptoms in most patients and can be used without localization of the leak. For cases that do not respond to blood patching, specialized spinal imaging is indicated to localize the site of the CSF leak, and several surgical and nonsurgical treatment options have become available, depending on the type of leak
(10). Dawwas GK, et al. Risk for recurrent venous thromboembolism and bleeding with apixaban compared with rivaroxaban: an analysis of real-world data. Ann Intern Med. 2021.
Apixaban and rivaroxaban are replacing vitamin K antagonists for the treatment of venous thromboembolism (VTE) in adults; however, head-to-head comparisons remain limited.
Objective: To assess the effectiveness and safety of apixaban compared with rivaroxaban in patients with VTE.
Design: Retrospective new-user cohort study.
Setting: U.S.-based commercial health care insurance database from 1 January 2015 to 30 June 2020.
Participants: Adults with VTE who were newly prescribed apixaban or rivaroxaban.
Measurements: The primary effectiveness outcome was recurrent VTE, a composite of deep venous thrombosis and pulmonary embolism. The primary safety outcome was a composite of gastrointestinal and intracranial bleeding.
Results: Of 49,900 eligible patients with VTE, 18,618 were new users of apixaban and 18,618 were new users of rivaroxaban. Median follow-up was 102 days (25th, 75th percentiles: 30, 128 days) among apixaban and 105 days (25th, 75th percentiles: 30, 140 days) among rivaroxaban users. After propensity score matching, apixaban (vs. rivaroxaban) was associated with a lower rate for recurrent VTE (hazard ratio, 0.77 [95% CI, 0.69 to 0.87]) and bleeding (hazard ratio, 0.60 [CI, 0.53 to 0.69]). The absolute reduction in the probability of recurrent VTE with apixaban versus rivaroxaban was 0.006 (CI, 0.005 to 0.011) within 2 months and 0.011 (CI, 0.011 to 0.013) within 6 months of initiation. The absolute reduction in the probability of gastrointestinal and intracranial bleeding with apixaban versus rivaroxaban was 0.011 (CI, 0.010 to 0.011) within 2 months and 0.015 (CI, 0.013 to 0.015) within 6 months of initiation.
Limitation: Short follow-up.
Conclusion: In this population-based cohort study, patients with VTE who were new users of apixaban had lower rates for recurrent VTE and bleeding than new users of rivaroxaban.