Journal scan: A review of 15 recent papers of immediate clinical significance, harvested from major international journals
From the desk of the Editor-in-Chief
(1). van Dyck CH., et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med. 2023;388:9-21
Background
The accumulation of soluble and insoluble aggregated amyloid-beta (Aβ) may initiate or potentiate pathologic processes in Alzheimer’s disease. Lecanemab, a humanized IgG1 monoclonal antibody that binds with high affinity to Aβ soluble protofibrils, is being tested in persons with early Alzheimer’s disease.
Methods
We conducted an 18-month, multicenter, double-blind, phase 3 trial involving persons 50 to 90 years of age with early Alzheimer’s disease (mild cognitive impairment or mild dementia due to Alzheimer’s disease) with evidence of amyloid on positron-emission tomography (PET) or by cerebrospinal fluid testing. Participants were randomly assigned in a 1:1 ratio to receive intravenous lecanemab (10 mg per kg of body weight every 2 weeks) or placebo. The primary end point was the change from baseline at 18 months in the score on the Clinical Dementia Rating-Sum of Boxes (CDR-SB; range, 0 to 18, with higher scores indicating greater impairment). Key secondary end points were the change in amyloid burden on PET, the score on the 14-item cognitive subscale of the Alzheimer’s Disease Assessment Scale (ADAS-cog14; range, 0 to 90; higher scores indicate greater impairment), the Alzheimer’s Disease Composite Score (ADCOMS; range, 0 to 1.97; higher scores indicate greater impairment), and the score on the Alzheimer’s Disease Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS-MCI-ADL; range, 0 to 53; lower scores indicate greater impairment).
Results
A total of 1795 participants were enrolled, with 898 assigned to receive lecanemab and 897 to receive placebo. The mean CDR-SB score at baseline was approximately 3.2 in both groups. The adjusted least-squares mean change from baseline at 18 months was 1.21 with lecanemab and 1.66 with placebo (difference, −0.45; 95% confidence interval [CI], −0.67 to −0.23; P<0.001). In a substudy involving 698 participants, there were greater reductions in brain amyloid burden with lecanemab than with placebo (difference, −59.1 centiloids; 95% CI, −62.6 to −55.6). Other mean differences between the two groups in the change from baseline favoring lecanemab were as follows: for the ADAS-cog14 score, −1.44 (95% CI, −2.27 to −0.61; P<0.001); for the ADCOMS, −0.050 (95% CI, −0.074 to −0.027; P<0.001); and for the ADCS-MCI-ADL score, 2.0 (95% CI, 1.2 to 2.8; P<0.001). Lecanemab resulted in infusion-related reactions in 26.4% of the participants and amyloid-related imaging abnormalities with edema or effusions in 12.6%.
Conclusions
Lecanemab reduced markers of amyloid in early Alzheimer’s disease and resulted in moderately less decline on measures of cognition and function than placebo at 18 months but was associated with adverse events. Longer trials are warranted to determine the efficacy and safety of lecanemab in early Alzheimer’s disease
(2). The first drug to slow the destruction of the brain in Alzheimer’s has been heralded as momentous.
The research breakthrough ends decades of failure and shows a new era of drugs to treat Alzheimer’s – the most common form of dementia – is possible.
Yet the medicine, lecanemab, has only a small effect and its impact on people’s daily lives is debated.
And the drug works in the early stages of the disease, so most would miss out without a revolution in spotting it.
Lecanemab attacks the sticky gunge – called beta amyloid – that builds up in the brains of people with Alzheimer’s.
For a medical field littered with duds, despair and disappointment, some see these trial results as a triumphant turning point.
Alzheimer’s Research UK said the findings were “momentous”.
One of the world’s leading researchers behind the whole idea of targeting amyloid 30 years ago, Prof John Hardy, said it was “historic” and was optimistic “we’re seeing the beginning of Alzheimer’s therapies”. Prof Tara Spires-Jones, from the University of Edinburgh, said the results were “a big deal because we’ve had a 100% failure rate for a long time”.
Which lifestyle changes can lower dementia risk?
How changes in nerve cells could offer protection in old age
Currently, people with Alzheimer’s are given other drugs to help manage their symptoms, but none change the course of the disease.
Lecanemab is an antibody – like those the body makes to attack viruses or bacteria – that has been engineered to tell the immune system to clear amyloid from the brain.
Amyloid is a protein that clumps together in the spaces between neurons in the brain and forms distinctive plaques that are one of the hallmarks of Alzheimer’s.
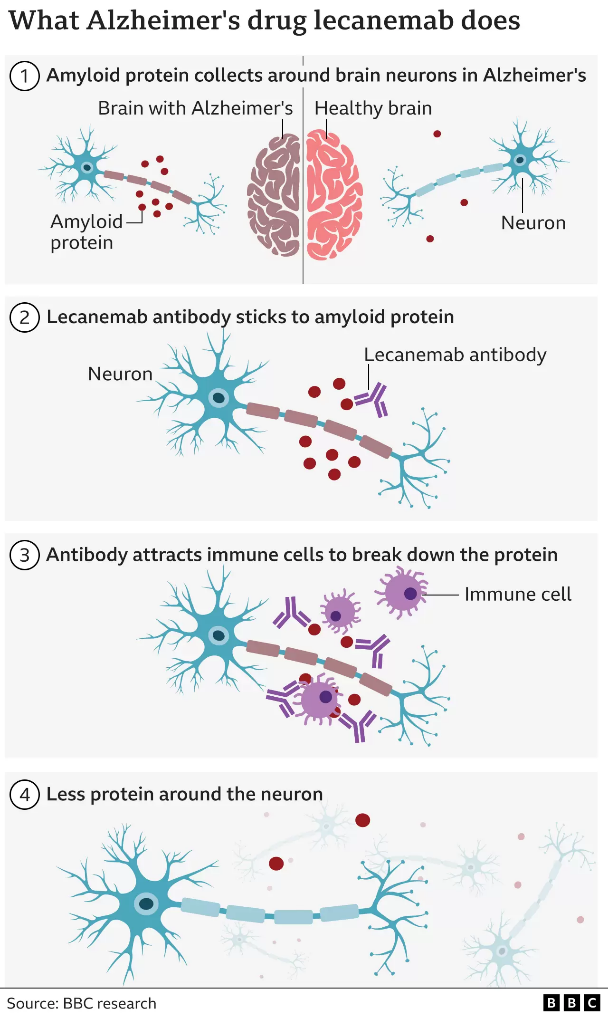
The large-scale trial involved 1,795 volunteers with early-stage Alzheimer’s. Infusions of lecanemab were given every fortnight.
The results, presented at the Clinical Trials on Alzheimer’s Disease conference in San Francisco and published in the New England Journal of Medicine, are not a miracle cure. The disease continued to rob people of their brain power, but that decline was slowed by around a quarter over the course of the 18 months of treatment.
The data is already being assessed by regulators in the US who will soon decide whether lecanemab can be approved for wider use. The developers – the pharmaceutical companies Eisai and Biogen – plan to begin the approval process in other countries next year.
David Essam, who is 78 and from Kent in the UK, took part in the international trial.
His Alzheimer’s meant he had to give up work as a joiner – he could no longer remember how to build a cabinet or use his tools. He now uses a digital watch as he can’t tell time using a clock face.
“He’s not the man he was, he needs help with most things, his memory in general is almost non-existent,” said his wife Cheryl. But she said the trial had given the family hope.
David said: “If somebody can slow it [Alzheimer’s] down and eventually stop it all together that would be brilliant, it’s just a horrible nasty thing.”
There are more than 55 million people in the world like David and the numbers with Alzheimer’s disease are projected to exceed 139 million by 2050.
Will it make a difference?
There is debate among scientists and doctors about the “real world” impact of lecanemab.
The slower decline with the drug was noticed using ratings of a person’s symptoms. It’s an 18-point scale, ranging from normal through to severe dementia. Those getting the drug were 0.45 points better off.
Prof Spires-Jones said that was a “small effect” on the disease, but “even though it is not dramatic, I would take it”.
Dr Susan Kohlhaas, from Alzheimer’s Research UK, said it was a “modest effect… but it gives us a little bit of a foothold” and the next generation of drugs would be better.
There are also risks. Brain scans showed a risk of brain bleeds (17% of participants) and brain swelling (13%). Overall, 7% of people given the drug had to stop because of side effects.
A crucial question is what happens after the 18 months of the trial, and the answers are still speculation.
Dr Elizabeth Coulthard, who treats patients at North Bristol NHS Trust, says that people have, on average, six years of living independently once mild cognitive impairment starts.
Slow that decline by a quarter and it could equate to an extra 19 months of independent life, “but we don’t know that yet”, she says.
It is even scientifically plausible that the effectiveness could be greater in longer trials. “I don’t think we can assume that this is it,” says Dr Kohlhass.
The emergence of drugs that do alter the course of the disease asks big questions of whether the health service is ready to use them.
The drugs have to be given early in the disease before too much damage to the brain is done, whereas most people referred to memory services are in the later stages of the disease.
That requires people coming forward at the earliest signs of memory problems and doctors being able to send them for amyloid tests – either brain scans or spinal fluid analysis – to a determine if they have Alzheimer’s or another form of dementia. At the moment only 1-2% of people with dementia have such tests.
“There’s an enormous gulf between current service provision and what we need to do, to deliver disease modifying therapies,” said Dr Coulthard.
She said that, currently, only those living near big medical centres or paying privately were likely to benefit.
Scientists also stressed that amyloid was only one part of the complex picture of Alzheimer’s disease and should not become the sole focus of therapies.
The immune system and inflammation are heavily involved in the disease and another toxic protein called tau is the one that’s found where brain cells are actually dying.
“That’s where I would put my money,” said Prof Spires-Jones.
She added: “I’m very excited we’re on the cusp of understanding enough to get a hold of the problem and we should have something that will make a bigger difference in a decade or so.”
Kate Lee, chief executive of Alzheimer’s Society charity, called for a 10-year government strategy on dementia to deal with what she called the “biggest health crisis we face in the UK”.
Speaking to Radio 4’s Today programme, she also said Lecanemab would not have a “huge impact” on those who already live with dementia.
But she added it should “make a big difference” for future generations.
(3). Avram Denburg. Essential anticancer medicines for children. Lancet 2022;23(12):P1479-1480.
Access to essential anticancer medicines is a vital component in the treatment of children with cancer. Inequities in such access are a reality both between and within countries, and constitute a key contributor to the disparate survival outcomes observed among children with cancer globally. 1 WHO has added anticancer medicines to its List of Essential Medicines for children (EMLc), to inform medicine selection, procurement, and supply management practices by national governments and health-care institutions internationally. 2 Given its aim to retain relevance across heterogeneous health systems, the WHO EMLc is most closely calibrated to system realities in low-income and middle-income countries, where the evolving epidemiological transition towards non-communicable diseases has precipitated an increasing childhood cancer burden and corollary needs for enhanced treatment capacities, including critical medicines.
(4). Markus P Schlaich et al. Dual endothelin antagonist aprocitentan for resistant hypertension (PRECISION): a multicentre, blinded, randomised, parallel-group, phase 3 trial. Lancet 2022;400(10367):P1927-1937.
Background
Resistant hypertension is associated with increased cardiovascular risk. The endothelin pathway has been implicated in the pathogenesis of hypertension, but it is currently not targeted therapeutically, thereby leaving this relevant pathophysiological pathway unopposed with currently available drugs. The aim of the study was to assess the blood pressure lowering efficacy of the dual endothelin antagonist aprocitentan in patients with resistant hypertension.
Methods
PRECISION was a multicentre, blinded, randomised, parallel-group, phase 3 study, which was done in hospitals or research centres in Europe, North America, Asia, and Australia. Patients were eligible for randomisation if their sitting systolic blood pressure was 140 mm Hg or higher despite taking standardised background therapy consisting of three antihypertensive drugs, including a diuretic. The study consisted of three sequential parts: part 1 was the 4-week double-blind, randomised, and placebo-controlled part, in which patients received aprocitentan 12·5 mg, aprocitentan 25 mg, or placebo in a 1:1:1 ratio; part 2 was a 32-week single (patient)-blind part, in which all patients received aprocitentan 25 mg; and part 3 was a 12-week double-blind, randomised, and placebo-controlled withdrawal part, in which patients were re-randomised to aprocitentan 25 mg or placebo in a 1:1 ratio. The primary and key secondary endpoints were changes in unattended office systolic blood pressure from baseline to week 4 and from withdrawal baseline to week 40, respectively. Secondary endpoints included 24-h ambulatory blood pressure changes. The study is registered on ClinicalTrials.gov, NCT03541174.
Findings
The PRECISION study was done from June 18, 2018, to April 25, 2022. 1965 individuals were screened and 730 were randomly assigned. Of these 730 patients, 704 (96%) completed part 1 of the study; of these, 613 (87%) completed part 2 and, of these, 577 (94%) completed part 3 of the study. The least square mean (SE) change in office systolic blood pressure at 4 weeks was -15·3 (SE 0·9) mm Hg for aprocitentan 12·5 mg, -15·2 (0·9) mm Hg for aprocitentan 25 mg, and -11·5 (0·9) mm Hg for placebo, for a difference versus placebo of -3·8 (1·3) mm Hg (97·5% CI -6·8 to -0·8, p=0·0042) and -3·7 (1·3) mm Hg (-6·7 to -0·8; p=0·0046), respectively. The respective difference for 24 h ambulatory systolic blood pressure was -4·2 mm Hg (95% CI -6·2 to -2·1) and -5·9 mm Hg (-7·9 to -3·8). After 4 weeks of withdrawal, office systolic blood pressure significantly increased with placebo versus aprocitentan (5·8 mm Hg, 95% CI 3·7 to 7·9, p<0·0001). The most frequent adverse event was mild-to-moderate oedema or fluid retention, occurring in 9%, 18%, and 2% for patients receiving aprocitentan 12·5 mg, 25 mg, and placebo, during the 4-week double-blind part, respectively. This event led to discontinuation in seven patients treated with aprocitentan. During the trial, a total of 11 treatment-emergent deaths occurred, none of which were regarded by the investigators to be related to study treatment.
Interpretation
In patients with resistant hypertension, aprocitentan was well tolerated and superior to placebo in lowering blood pressure at week 4 with a sustained effect at week 40.
(5). Mebazaa A. Safety, tolerability and efficacy of up-titration of guideline-directed medical therapies for acute heart failure (STRONG-HF): a multinational, open-label, randomised, trial. Lancet 2022;400(10367):P1938-1952.
Background
There is a paucity of evidence for dose and pace of up-titration of guideline-directed medical therapies after admission to hospital for acute heart failure.
Methods
In this multinational, open-label, randomised, parallel-group trial (STRONG-HF), patients aged 18-85 years admitted to hospital with acute heart failure, not treated with full doses of guideline-directed drug treatment, were recruited from 87 hospitals in 14 countries. Before discharge, eligible patients were randomly assigned (1:1), stratified by left ventricular ejection fraction (≤40% vs >40%) and country, with blocks of size 30 within strata and randomly ordered sub-blocks of 2, 4, and 6, to either usual care or high-intensity care. Usual care followed usual local practice, and high-intensity care involved the up-titration of treatments to 100% of recommended doses within 2 weeks of discharge and four scheduled outpatient visits over the 2 months after discharge that closely monitored clinical status, laboratory values, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) concentrations. The primary endpoint was 180-day readmission to hospital due to heart failure or all-cause death. Efficacy and safety were assessed in the intention-to-treat (ITT) population (ie, all patients validly randomly assigned to treatment). The primary endpoint was assessed in all patients enrolled at hospitals that followed up patients to day 180. Because of a protocol amendment to the primary endpoint, the results of patients enrolled on or before this amendment were down-weighted. This study is registered with ClinicalTrials.gov, NCT03412201, and is now complete.
Findings
Between May 10, 2018, and Sept 23, 2022, 1641 patients were screened and 1078 were successfully randomly assigned to high-intensity care (n=542) or usual care (n=536; ITT population). Mean age was 63·0 years (SD 13·6), 416 (39%) of 1078 patients were female, 662 (61%) were male, 832 (77%) were White or Caucasian, 230 (21%) were Black, 12 (1%) were other races, one (<1%) was Native American, and one (<1%) was Pacific Islander (two [<1%] had missing data on race). The study was stopped early per the data and safety monitoring board’s recommendation because of greater than expected between-group differences. As of data cutoff (Oct 13, 2022), by day 90, a higher proportion of patients in the high-intensity care group had been up-titrated to full doses of prescribed drugs (renin-angiotensin blockers 278 [55%] of 505 vs 11 [2%] of 497; β blockers 249 [49%] vs 20 [4%]; and mineralocorticoid receptor antagonists 423 [84%] vs 231 [46%]). By day 90, blood pressure, pulse, New York Heart Association class, bodyweight, and NT-proBNP concentration had decreased more in the high-intensity care group than in the usual care group. Heart failure readmission or all-cause death up to day 180 occurred in 74 (15·2% down-weighted adjusted Kaplan-Meier estimate) of 506 patients in the high-intensity care group and 109 (23·3%) of 502 patients in the usual care group (adjusted risk difference 8·1% [95% CI 2·9-13·2]; p=0·0021; risk ratio 0·66 [95% CI 0·50-0·86]). More adverse events by 90 days occurred in the high-intensity care group (223 [41%] of 542) than in the usual care group (158 [29%] of 536) but similar incidences of serious adverse events (88 [16%] vs 92 [17%]) and fatal adverse events (25 [5%] vs 32 [6%]) were reported in each group.
Interpretation
An intensive treatment strategy of rapid up-titration of guideline-directed medication and close follow-up after an acute heart failure admission was readily accepted by patients because it reduced symptoms, improved quality of life, and reduced the risk of 180-day all-cause death or heart failure readmission compared with usual care.
(6). Amit K. Sahu. Filarial Dance Sign in Lymphatic Filariasis of the Scrotum. N Engl J Med. 2022;387:e61.
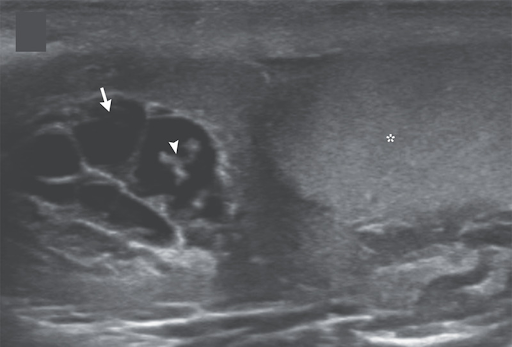
A 26-year-old man presented to the outpatient clinic with a 1-month history of pain and swelling in the scrotum and low-grade fevers. His body temperature was 37.5°C. On examination, there was tenderness and swelling of the right side of the scrotum. Laboratory studies showed an absolute eosinophil count of 1180 per cubic millimeter (reference value, <500). An ultrasound examination of the scrotum showed anechoic tubular channels (arrow) near the right testis (asterisk) and epididymal head. Color flow was absent in the channels on color-flow Doppler imaging. Echogenic, linear structures could be seen moving within one of the channels (arrowhead; see video), a finding in lymphatic filariasis known as “filarial dance sign.” Lymphatic filariasis is a parasitic infection caused by mosquito-transmitted nematodes. The dance sign represents the undulations of live worms that have migrated into lymphatic channels, causing dilation and dysfunction of the channels. Ultrasound-guided aspiration of scrotal lymphatic fluid was performed, and live microfilariae of the species Wuchereria bancrofti were identified on light microscopy. The organisms were also seen on a peripheral-blood smear. After a 3-week course of oral diethylcarbamazine, the patient’s symptoms abated, and the motile microfilariae were no longer seen on ultrasonography.
(7). Areef Ishani et al. Chlorthalidone vs. Hydrochlorothiazide for Hypertension-Cardiovascular Events. 2022
Background
Whether chlorthalidone is superior to hydrochlorothiazide for preventing major adverse cardiovascular events in patients with hypertension is unclear.
Methods
In a pragmatic trial, we randomly assigned adults 65 years of age or older who were patients in the Department of Veterans Affairs health system and had been receiving hydrochlorothiazide at a daily dose of 25 or 50 mg to continue therapy with hydrochlorothiazide or to switch to chlorthalidone at a daily dose of 12.5 or 25 mg. The primary outcome was a composite of nonfatal myocardial infarction, stroke, heart failure resulting in hospitalization, urgent coronary revascularization for unstable angina, and non-cancer-related death. Safety was also assessed.
Results
A total of 13,523 patients underwent randomization. The mean age was 72 years. At baseline, hydrochlorothiazide at a dose of 25 mg per day had been prescribed in 12,781 patients (94.5%). The mean baseline systolic blood pressure in each group was 139 mm Hg. At a median follow-up of 2.4 years, there was little difference in the occurrence of primary-outcome events between the chlorthalidone group (702 patients [10.4%]) and the hydrochlorothiazide group (675 patients [10.0%]) (hazard ratio, 1.04; 95% confidence interval, 0.94 to 1.16; P=0.45). There were no between-group differences in the occurrence of any of the components of the primary outcome. The incidence of hypokalemia was higher in the chlorthalidone group than in the hydrochlorothiazide group (6.0% vs. 4.4%, P<0.001).
Conclusions
In this large pragmatic trial of thiazide diuretics at doses commonly used in clinical practice, patients who received chlorthalidone did not have a lower occurrence of major cardiovascular outcome events or non-cancer-related deaths than patients who received hydrochlorothiazide.
(8). PREVAC Study Team. Randomized Trial of Vaccines for Zaire Ebola Virus Disease. 2022
Background
Questions remain concerning the rapidity of immune responses and the durability and safety of vaccines used to prevent Zaire Ebola virus disease.
Methods
We conducted two randomized, placebo-controlled trials — one involving adults and one involving children — to evaluate the safety and immune responses of three vaccine regimens against Zaire Ebola virus disease: Ad26.ZEBOV followed by MVA-BN-Filo 56 days later (the Ad26-MVA group), rVSVΔG-ZEBOV-GP followed by placebo 56 days later (the rVSV group), and rVSVΔG-ZEBOV-GP followed by rVSVΔG-ZEBOV-GP 56 days later (the rVSV-booster group). The primary end point was antibody response at 12 months, defined as having both a 12-month antibody concentration of at least 200 enzyme-linked immunosorbent assay units (EU) per milliliter and an increase from baseline in the antibody concentration by at least a factor of 4.
Results
A total of 1400 adults and 1401 children underwent randomization. Among both adults and children, the incidence of injection-site reactions and symptoms (e.g., feverishness and headache) was higher in the week after receipt of the primary and second or booster vaccinations than after receipt of placebo but not at later time points. These events were largely low-grade. At month 12, a total of 41% of adults (titer, 401 EU per milliliter) and 78% of children (titer, 828 EU per milliliter) had a response in the Ad26-MVA group; 76% (titer, 992 EU per milliliter) and 87% (titer, 1415 EU per milliliter), respectively, had a response in the rVSV group; 81% (titer, 1037 EU per milliliter) and 93% (titer, 1745 EU per milliliter), respectively, had a response in the rVSV-booster group; and 3% (titer, 93 EU per milliliter) and 4% (titer, 67 EU per milliliter), respectively, had a response in the placebo group (P<0.001 for all comparisons of vaccine with placebo). In both adults and children, antibody responses with vaccine differed from those with placebo beginning on day 14.
Conclusions
No safety concerns were identified in this trial. With all three vaccine regimens, immune responses were seen from day 14 through month 12.
(9). Daniel Weghuber et al. Once-Weekly Semaglutide in Adolescents with Obesity. N Engl J Med 2022;387:2245-2257
Background
A once-weekly, 2.4-mg dose of subcutaneous semaglutide, a glucagon-like peptide-1 receptor agonist, is used to treat obesity in adults, but assessment of the drug in adolescents has been lacking.
Methods
In this double-blind, parallel-group, randomized, placebo-controlled trial, we enrolled adolescents (12 to <18 years of age) with obesity (a body-mass index [BMI] in the 95th percentile or higher) or with overweight (a BMI in the 85th percentile or higher) and at least one weight-related coexisting condition. Participants were randomly assigned in a 2:1 ratio to receive once-weekly subcutaneous semaglutide (at a dose of 2.4 mg) or placebo for 68 weeks, plus lifestyle intervention. The primary end point was the percentage change in BMI from baseline to week 68; the secondary confirmatory end point was weight loss of at least 5% at week 68.
Results
A total of 201 participants underwent randomization, and 180 (90%) completed treatment. All but one of the participants had obesity. The mean change in BMI from baseline to week 68 was −16.1% with semaglutide and 0.6% with placebo (estimated difference, −16.7 percentage points; 95% confidence interval [CI], −20.3 to −13.2; P<0.001). At week 68, a total of 95 of 131 participants (73%) in the semaglutide group had weight loss of 5% or more, as compared with 11 of 62 participants (18%) in the placebo group (estimated odds ratio, 14.0; 95% CI, 6.3 to 31.0; P<0.001). Reductions in body weight and improvement with respect to cardiometabolic risk factors (waist circumference and levels of glycated hemoglobin, lipids [except high-density lipoprotein cholesterol], and alanine aminotransferase) were greater with semaglutide than with placebo. The incidence of gastrointestinal adverse events was greater with semaglutide than with placebo (62% vs. 42%). Five participants (4%) in the semaglutide group and no participants in the placebo group had cholelithiasis. Serious adverse events were reported in 15 of 133 participants (11%) in the semaglutide group and in 6 of 67 participants (9%) in the placebo group.
Conclusions
Among adolescents with obesity, once-weekly treatment with a 2.4-mg dose of semaglutide plus lifestyle intervention resulted in a greater reduction in BMI than lifestyle intervention alone.
(10). Baby’s life ‘probably saved’ by umbilical stem cells
A heart surgeon says he “probably saved the life” of a baby by carrying out a “world-first” operation using stem cells from placentas.
Professor Massimo Caputo from the Bristol Heart Institute used pioneering stem cell “scaffolding” to correct baby Finley’s heart defect.
He hopes to develop the technology so children born with congenital cardiac disease won’t need as many operations.
But he was born with the main arteries in his heart the wrong way round and at just four days old had his first open-heart surgery at Bristol Royal Hospital for Children.
Unfortunately the surgery didn’t solve the problem and his heart function deteriorated significantly, with the left side of the heart suffering from a severe lack of blood flow.
After weeks in intensive care it looked like there was no conventional way to treat Finley’s condition and he was reliant on drugs to keep his heart going.
But a new procedure was tried, involving stem cells from a placenta bank.
Prof Caputo injected the cells directly into Finley’s heart in the hope they would help damaged blood vessels grow.
The so-called “allogeneic” cells were grown by scientists at the Royal Free Hospital in London, and millions of them were injected into Finley’s heart muscle.
Allogeneic cells have the ability to grow into tissue that is not rejected and in Finley’s case, have regenerated damaged heart muscle.
“We weaned him from all the drugs he was on, we weaned him from ventilation,” said Prof Caputo.
“He was discharged from ITU and is now a happy growing little boy.”
Using a bio-printer, a stem cell scaffold is made to repair abnormalities to valves in blood vessels, and to mend holes between the two main pumping chambers of the heart.
Artificial tissue is normally used used on babies for cardiac repairs, but it can fail and it doesn’t grow with the heart, so as the children grow, they require more operations.
Prof Caputo hopes a clinical trial on the patches will happen in the next two years, after successful laboratory work.
every day in the UK, around 13 babies are diagnosed with a congenital heart defect – a heart condition that develops before the baby is born, according to the British Heart Foundation.
Because the materials used to fix the heart can be rejected by the patient’s immune system, they can cause scarring in the heart that can lead to other complications, and can gradually break down and fail in just a few months or years.
A child might therefore have to go through the same heart operation multiple times throughout its childhood- around 200 repeat operations for congenital heart defects are carried out every year in the UK.
Prof Caputo and his team say the stem cell technology could save the NHS an estimated £30,000 for every operation no longer needed, saving millions of pounds each year.
Dr Stephen Minger, an expert in stem cell biology and director of SLM Blue Skies Innovations Ltd, applauded the research.
He said: “Most studies that I am aware of in adults with heart dysfunction or failure show only minimal therapeutic benefit with stem cell infusion.
“I’m happy that the clinical team will go on to do a standard clinical trial which should tell us if this was a ‘one-off’ success and also give us some better understanding of mechanisms behind this.”
(11). Safi U Khan, et al. Statin related muscle symptoms: is it time to move on. BMJ 2022;379:o2939
Fresh evidence adds confidence to shared decision making about statins
Inhibitors of hydroxymethylglutaryl coenzyme A reductase (statins) are among the most widely investigated and prescribed drugs in cardiovascular medicine, and they have substantial effects on the outcomes of atherosclerotic cardiovascular disease.12 Leading guidelines from US and Europe2 broadly recommend statins for people with high low density lipoprotein (LDL) cholesterol concentrations and a high risk of cardiovascular events, including those with a history of cardiovascular disease (secondary prevention).12
The approach for people at lower risk (including for primary prevention) is more variable. The European Society of Cardiology recommends statins for patients with high LDL cholesterol concentrations, even when their overall risk of cardiovascular events is low (<1% over 10 years),2 whereas the American College of Cardiology and American Heart Association recommend lifestyle modifications not statins for people with a risk below 5% over 10 years.1
Despite clear indications, some studies show that roughly half of patients stop taking statins after six months, and only a quarter of patients with high cardiovascular risk continue with treatment long term.34 Statin associated muscle symptoms, described as myalgia, cramping, or fatigue, are the most common cause of statin intolerance.
Landmark trials of statins reported low frequencies of statin associated myalgia.56 But concerns remained about the possibility of under-reporting or underestimations because participants with comorbidities, who are more likely to develop these symptoms, were excluded.7 A more recent participant level meta-analysis of 19 placebo controlled trials of statin therapy, including more than 33 000 patients (48% with atherosclerotic cardiovascular disease) attempted to overcome these shortcomings.7 The authors found that, compared with placebo, statin treatment was associated with only 11 excess incidents of muscle pain or weakness per 1000 person years of treatment during the first year.7 They found no significant difference in absolute risk between statin and placebo groups after one year.
Myalgia was common among participants, regardless of statin use.7 The proportion of patients reporting muscle pain or weakness was 27.1% in the statin arm and 26.6% in the placebo arm. These event rates remained consistent across subgroups stratified by age, sex, ethnicity, history of vascular disease, diabetes, body mass index, LDL cholesterol concentration, and estimated glomerular filtration rate. The authors calculated that only 1 in 15 reports of muscle symptoms in statin groups were attributable to statins.7 Furthermore, the additional absolute risk of such events remained low across different intensities of therapy.7 Creatinine kinase activity, a more objective outcome than patient reported symptoms, was increased only minimally with statin therapy.
Excess risk of muscle pain or weakness was evident only during the first year, while evidence suggests that the benefits of LDL lowering therapies, including statins, accumulate over longer treatment durations.8
Findings from the new meta-analysis7 of muscle symptoms should be considered in the context of several limitations, including heterogeneity in the methods used to ascertain muscle symptoms; uncertainties about the nature of symptoms among participants who stopped their statin compared with those who continued; and variable outcome definitions, although the definitions probably reflect the various ways patients describe their symptom. The authors had limited data on whether muscle symptoms led to the discontinuation of allocated treatment, and participants with statin related myalgia dropping out might partly explain the lack of risk difference after one year. Data were also limited on comorbidities such as hypothyroidism and concomitant medications that might cause or influence muscle symptoms.7
Accurate estimation of the risk-benefit balance associated with statins is particularly important for primary prevention. In a recent meta-analysis of 62 primary prevention trials (120 456 patients; <3% risk of major cardiovascular events at average follow-up 3.9 years), statins were associated with 15 events of self-reported muscle symptoms per 10 000 patients a year compared with a non-statin control.9 But statins were also associated with a significant reduction in myocardial infarction (19 fewer per 10 000), stroke (9 fewer), and cardiovascular deaths (8 fewer) compared with controls.
Individuals making treatment decisions often have to rely on clinical trials reporting average treatment effects across large sample sizes, which may not reflect their own personal balance of benefits and harms.10 To help bridge this gap, the British Heart Foundation conducted an n-of-1 trial (Samson), which used a crossover design to assess the tolerability of statin, placebo, or no treatment among 60 patients with a history of statin intolerance, most of whom had a history of statin associated muscle symptoms (85%) and a 10 year risk of cardiovascular events <25% (77%).11
Participants were randomised to one month periods of atorvastatin 20 mg, placebo, and no treatment for a year. Mean symptom intensity on a 100 point scale— recorded daily—was 8.0 during periods of no treatment compared with 15.4 when taking a placebo and 16.3 taking a statin (P=0.38). The ratio of symptom intensity induced by a placebo to the symptom intensity induced by a statin (nocebo ratio) was 0.9, suggesting that 90% of the symptom burden induced by a statin challenge was also evoked by a placebo.12 In other words, even the possibility of receiving a statin versus the certainty of not receiving one was enough to induce the sensation of muscle pain. Another n-of-1 trial from the UK in 151 participants with a history of statin intolerancealso found no effect of statins on muscle symptoms.13
While further n-of-1 trials may help to clarify the nature and severity of reported myalgia, clear evidence indicates that statin associated muscle symptoms are uncommon and that most are not related to muscle damage.14
While clinicians must respect patients’ beliefs and preferences, they must also offer treatment when evidence shows the potential for substantial benefits, particularly in avoiding catastrophic events. Combining these obligations requires meaningful shared decision making about statins, including acknowledgment of patients’ concerns and a clear, personalised, and evidence based discussion of the likely benefits and harms of treatment. We now have much better evidence to approach these conversations confidently.
(12). Lars M. Asmis. Recombinant ADAMTS13 for Hereditary Thrombotic Thrombocytopenic Purpura. N Engl J Med. 2022;387:2356-2361.
Summary
A 27-year-old patient with a history of severe obstetrical complications and arterial thrombosis received a diagnosis of hereditary thrombotic thrombocytopenic purpura (TTP) due to severe ADAMTS13 deficiency when she presented with an acute episode in the 30th week of her second pregnancy. When the acute episode of hereditary TTP became plasma-refractory and fetal death was imminent, weekly injections of recombinant ADAMTS13 at a dose of 40 U per kilogram of body weight were initiated. The patient’s platelet count normalized, and the growth of the fetus stabilized. At 37 weeks 1 day of gestation, a small-for-gestational-age boy was delivered by cesarean section. At the time of this report, the patient and her son were well, and she continued to receive injections of recombinant ADAMTS13 every 2 weeks.
(13). Erin R Whitehouse. Human rabies despite post-exposure prophylaxis: a systematic review of fatal breakthrough infections after zoonotic exposures. 2022
Summary
Post-exposure prophylaxis (PEP) for rabies is widely administered and highly effective. Nevertheless, sporadic breakthrough infections (ie, rabies in people who have started PEP) have been reported. We conducted a systematic review of articles published between Jan 1, 1980 and June 1, 2022 to characterise breakthrough infections. After reviewing 3380 articles from across all continents, we identified 52 articles, which included a total of 122 breakthrough infections. We classified breakthrough infections on the basis of adherence to core practices (ie, wound cleaning and vaccine administration). Of 86 breakthrough infections with data, median time from exposure to symptom onset was 20 days (IQR 16-24). Most (89 [77%] of 115) participants received PEP within 2 days of an exposure. Severe wounds (defined as those involving multiple wound sites or bites to the head, face, or neck) were common (80 [69%] of 116 [with data]). Deviations from core practices were reported in 68 (56%) of 122 cases. Other possible causes for breakthrough infections included errors in the administration of rabies immunoglobulin, delays in seeking health care, and comorbidities or immunosuppression. Cold-chain integrity assessments and potency testing of PEP biologics were only rarely assessed (8 [7%] of 122 cases), neither of which were found to be a cause of breakthrough infections. Timely and appropriate administration of PEP is crucial to prevent rabies, and although people with high-risk exposures or immunosuppression can develop rabies despite adherence to core practices, this occurrence remains exceedingly rare.
(14). Bern-Thomas Nyang’wa. A 24-Week, All-Oral Regimen for Rifampin-Resistant Tuberculosis. N Engl J Med 2022; 387:2331-2343.
Background
In patients with rifampin-resistant tuberculosis, all-oral treatment regimens that are more effective, shorter, and have a more acceptable side-effect profile than current regimens are needed.
Methods
We conducted an open-label, phase 2-3, multicenter, randomized, controlled, noninferiority trial to evaluate the efficacy and safety of three 24-week, all-oral regimens for the treatment of rifampin-resistant tuberculosis. Patients in Belarus, South Africa, and Uzbekistan who were 15 years of age or older and had rifampin-resistant pulmonary tuberculosis were enrolled. In stage 2 of the trial, a 24-week regimen of bedaquiline, pretomanid, linezolid, and moxifloxacin (BPaLM) was compared with a 9-to-20-month standard-care regimen. The primary outcome was an unfavorable status (a composite of death, treatment failure, treatment discontinuation, loss to follow-up, or recurrence of tuberculosis) at 72 weeks after randomization. The noninferiority margin was 12 percentage points.
Results
Recruitment was terminated early. Of 301 patients in stage 2 of the trial, 145, 128, and 90 patients were evaluable in the intention-to-treat, modified intention-to-treat, and per-protocol populations, respectively. In the modified intention-to-treat analysis, 11% of the patients in the BPaLM group and 48% of those in the standard-care group had a primary-outcome event (risk difference, −37 percentage points; 96.6% confidence interval [CI], −53 to −22). In the per-protocol analysis, 4% of the patients in the BPaLM group and 12% of those in the standard-care group had a primary-outcome event (risk difference, −9 percentage points; 96.6% CI, −22 to 4). In the as-treated population, the incidence of adverse events of grade 3 or higher or serious adverse events was lower in the BPaLM group than in the standard-care group (19% vs. 59%).
Conclusions
In patients with rifampin-resistant pulmonary tuberculosis, a 24-week, all-oral regimen was noninferior to the accepted standard-care treatment, and it had a better safety profile.
(15). Evan S. Dellon, et al. Dupilumab in Adults and Adolescents with Eosinophilic Esophagitis. N Engl J Med. 2022
Background
Dupilumab, a fully human monoclonal antibody, blocks interleukin-4 and interleukin-13 signaling, which have key roles in eosinophilic esophagitis.
Methods
We conducted a three-part, phase 3 trial in which patients 12 years of age or older underwent randomization in a 1:1 ratio to receive subcutaneous dupilumab at a weekly dose of 300 mg or placebo (Part A) or in a 1:1:1 ratio to receive 300 mg of dupilumab either weekly or every 2 weeks or weekly placebo (Part B) up to week 24. Eligible patients who completed Part A or Part B continued the trial in Part C, in which those who completed Part A received dupilumab at a weekly dose of 300 mg up to week 52 (the Part A-C group); Part C that included the eligible patients from Part B is ongoing. The two primary end points at week 24 were histologic remission (≤6 eosinophils per high-power field) and the change from baseline in the Dysphagia Symptom Questionnaire (DSQ) score (range, 0 to 84, with higher values indicating more frequent or more severe dysphagia).
Results
In Part A, histologic remission occurred in 25 of 42 patients (60%) who received weekly dupilumab and in 2 of 39 patients (5%) who received placebo (difference, 55 percentage points; 95% confidence interval [CI], 40 to 71; P<0.001). In Part B, histologic remission occurred in 47 of 80 patients (59%) with weekly dupilumab, in 49 of 81 patients (60%) with dupilumab every 2 weeks, and in 5 of 79 patients (6%) with placebo (difference between weekly dupilumab and placebo, 54 percentage points; 95% CI, 41 to 66 [P<0.001]; difference between dupilumab every 2 weeks and placebo, 56 percentage points; 95% CI, 43 to 69 [not significant per hierarchical testing]). The mean (±SD) DSQ scores at baseline were 33.6±12.41 in Part A and 36.7±11.22 in Part B; the scores improved with weekly dupilumab as compared with placebo, with differences of -12.32 (95% CI, -19.11 to -5.54) in Part A and -9.92 (95% CI, -14.81 to -5.02) in Part B (both P<0.001) but not with dupilumab every 2 weeks (difference in Part B, -0.51; 95% CI, -5.42 to 4.41). Serious adverse events occurred in 9 patients during the Part A or B treatment period (in 7 who received weekly dupilumab, 1 who received dupilumab every 2 weeks, and 1 who received placebo) and in 1 patient in the Part A-C group during the Part C treatment period who received placebo in Part A and weekly dupilumab in Part C.
Conclusions
Among patients with eosinophilic esophagitis, subcutaneous dupilumab administered weekly improved histologic outcomes and alleviated symptoms of the disease.